
無機化學/配位化學與晶體場理論導論

配位化合物(或配合物)是包含過渡金屬離子與一個或多個配體之間鍵的分子和擴充套件固體。在形成這些配位共價鍵時,金屬離子充當路易斯酸,配體充當路易斯鹼。通常,配體具有孤對電子,並且鍵的形成是透過包含此電子對的分子軌道與金屬離子的d軌道重疊而形成的。在配位配合物中常見的配體是中性分子(H2O、NH3、吡啶等有機鹼、CO、NO、H2、乙烯和膦PR3)和陰離子(鹵化物、CN-、SCN-、環戊二烯(C5H5-)、H-等)。所得配合物可以是陽離子(例如,[Cu(NH3)4]2+)、中性([Pt(NH3)2Cl2])或陰離子([Fe(CN)6]4-)。正如我們將在下面看到的,作為布朗斯臺德鹼具有弱或可忽略不計強度的配體(例如,CO、CN-、H2O和Cl-)仍然可以在形成過渡金屬配合物中成為有效的路易斯鹼。
對於作為路易斯鹼的配體,配位共價鍵(也稱為配位鍵)通常被畫成線,或者有時用箭頭表示電子對“屬於”配體X。

在計算金屬上的電子(如下所述)時,慣例是將配位鍵中的兩個電子都分配給配體,儘管實際上鍵通常是極性共價鍵,並且電子在金屬和配體之間共享。
在寫出配位化合物的分子式時,我們使用方括號[...]括起來直接鍵合在一起的金屬離子和配體。因此,化合物[Co(NH3)5Cl]Cl2包含八面體[Co(NH3)5Cl]2+離子,其中五個氨分子和一個氯離子直接鍵合到金屬上,以及兩個未與金屬配位的Cl-陰離子。
.png)

歷史。配位化合物已經存在了幾個世紀,但最初人們並不瞭解它們的結構。例如,普魯士藍,其經驗式為Fe7(CN)18•xH2O,是一種不溶性的深藍色固體,自1704年迪斯巴赫偶然發現以來一直用作顏料。普魯士藍實際上包含Fe3+陽離子和[Fe(CN)6]4-陰離子,更具描述性的表述為(Fe3+)4([Fe(CN)6]4-)3•xH2O。像Co3+的氨配合物這樣的簡單化合物是化學家所熟知的,但它們不符合離子固體的預期行為。例如,氯化鈷(III)六氨,[Co(NH3)6]Cl3被配製為CoCl3•6NH3。它具有神秘的特性,因為它像離子固體一樣溶於水中,但在重結晶時保留了六個氨分子。更有趣的是,觀察到過渡金屬配合物(如[Co(NH3)4Cl2]Cl)可以製成化學上不同的形式(異構體)。這個難題由阿爾弗雷德·維爾納解決,他於1893年提出這些Co配合物包含八面體配位的金屬離子,這些金屬離子與六個配體形成主要(共價)鍵。維爾納透過電導率測量表明,CoCl3•6NH3溶液每摩爾含有三個遊離Cl-陰離子和一個[Co(NH3)6]3+陽離子。後來的磁化率測量證實了鹽及其溶液中都存在抗磁性Co3+。維爾納的理論也解釋了[Co(NH3)4Cl2]+存在兩種(且僅兩種)結構異構體的原因。
像有機化合物一樣,過渡金屬配合物的大小、形狀、電荷和穩定性可以有很大的差異。我們將看到由金屬的d軌道形成的鍵在很大程度上控制了這些性質。
第5章的學習目標
- 確定配合物中過渡金屬的氧化態並分配d電子數。
- 推匯出八面體、拉長的八面體、四方錐體、平面正方形和四面體配合物的d軌道分裂模式。
- 對於八面體和四面體配合物,確定未成對電子的數量並計算晶體場穩定化能。
- 瞭解分光化學序列,解釋不同型別的配體如何影響晶體場分裂能,並用它來預測高自旋與低自旋配合物以及過渡金屬配合物的顏色。
- 利用過渡金屬配合物的磁矩確定其自旋狀態。
- 瞭解Jahn-Teller效應的起源及其對配合物形狀、顏色和反應性的影響。
- 瞭解螯合和環狀配體形成的配合物的額外穩定性。
d軌道是過渡金屬配合物的邊界軌道(HOMO和LUMO)。配合物的許多重要性質——其形狀、顏色、磁性和反應性——取決於金屬d軌道的電子佔據情況。為了理解和解釋這些性質,瞭解如何計算d電子非常重要。

因為過渡金屬通常比形成金屬-配體鍵的配體上的原子(C、N、O、Cl、P...)電負性更低,所以我們的慣例是將鍵中的兩個電子都分配給配體。例如,在鐵氰根配合物[Fe(CN)6]3-中,如果氰化物配體保留了它的兩個電子,則將其配製為CN-。透過差值,鐵必須是Fe3+,因為電荷(3+ + 6(1-))必須加起來等於配合物的總電荷-3。
下一步是確定Fe3+離子有多少個d電子。規則是將鐵的所有價電子都算作d電子。鐵位於第8族,因此
- 第8族 - 3+電荷 = d5(或3d5)
- 8 - 3 = 5
同樣的過程可以應用於任何過渡金屬配合物。例如,考慮配合物[Cu(NH3)4]2+。由於氨是中性配體,因此Cu處於2+氧化態。銅(II)位於元素週期表第11族,其價電子層中有11個電子,減去2,剩下9個d電子(3d9)。在中性配合物[Rh(OH)3(H2O)3]中,Rh處於+3氧化態,位於第9族,因此電子數為4d6。第12族的鋅(II)在[Zn(NH3)4]2+中將有10個d電子,形成一個完整的電子層,而錳(VII)在MnO4-中則沒有d電子。鎳羰基Ni(CO)4含有中性CO配體和處於零氧化態的Ni。由於Ni位於第10族,我們將Ni上的電子數記為3d10。
關於電子計數的一個常見困惑是金屬上s電子的去向。例如,我們的電子計數規則預測Ti在八面體配合物[Ti(H2O)6]3+中為3d1。但根據構造原理,自由Ti原子的電子構型為4s23d2。為什麼Ti3+離子是3d1而不是4s1?同樣,為什麼我們將Mn2+分配為3d5而不是4s23d3?簡而言之,是因為在金屬配合物中,金屬s軌道的能量高於自由原子中的能量,因為它們具有反鍵特性。我們將在第5.2節中使用分子軌道圖來證明這一說法。
共價鍵分類(CBC)方法。儘管我們上面開發的電子計數規則很有用,並且適用於所有型別的配合物,但將配合物中所有共享電子分配給配體的做法並不總是代表真實的鍵合影像。這種影像在配體電負性遠大於金屬的情況下最為準確。但實際上,存在各種各樣的配體,包括H、烷基、環戊二烯基等,其中金屬和配體的電負性相當。在這些情況下,特別是對於電負性相對較高的後過渡金屬,我們應該將金屬-配體鍵視為共價鍵。CBC方法,也稱為LXZ符號,由M. L. H. Green[2]於1995年提出,以便更好地描述不同型別的金屬-配體鍵。下面的分子軌道圖總結了L、X和Z配體之間的區別。[3]其中,L和X是最常見的型別。

L型配體是路易斯鹼,無論使用何種電子計數方法,都會向金屬中心貢獻兩個電子。這些電子可以來自孤對電子、π或σ供體。這些配體與金屬之間形成的鍵是配位共價鍵,也稱為配位鍵。此類配體的例子包括CO、PR3、NH3、H2O、卡賓(=CRR')和烯烴。


X型配體是指在使用中性配體電子計數法時,向金屬貢獻一個電子並從金屬接受一個電子,或者在使用供體對電子計數法時,向金屬貢獻兩個電子的配體。[4]無論將其視為中性還是陰離子,這些配體都會產生正常的共價鍵。此類配體的一些例子包括H、CH3、鹵素和NO(彎曲)。
Z型配體是指從金屬中心接受兩個電子,而不是像其他兩種型別的配體那樣進行供體。然而,這些配體也像L型一樣形成配位共價鍵。這種型別的配體通常不使用,因為在某些情況下它可以用L和X表示。例如,如果Z配體伴隨著L型,它可以寫成X2。這些配體的例子包括路易斯酸,例如BR3。
一些多齒配體可以作為配體型別的組合。一個著名的例子是環戊二烯基(或Cp)配體,C5H5。我們將這種中性配體分類為[L2X],其中兩個L官能團對應於兩個“烯烴”片段,而X官能團對應於環中CH“自由基”碳。新增一個電子會生成Cp-陰離子,它具有六個π電子,因此是平面的和芳香性的。在二茂鐵配合物Cp2Fe中,使用“標準”供體對計數方法,我們可以將兩個Cp-配體分別視為具有六個π電子,根據差值,Fe處於+2氧化態。Fe2+離子為d6。因此,配合物中的鐵原子(無論使用何種計數方法)在其配位環境中都有6+6+6=18個電子,對於過渡金屬配合物來說,這是一個特別穩定的電子數。
晶體場理論是解釋過渡金屬配合物的結構和性質的最簡單的模型之一。該理論基於金屬-配體相互作用的靜電學,因此,當金屬-配體鍵基本上是共價鍵時,其結果僅是近似的。但由於該模型有效地利用了分子對稱性,因此在描述金屬配合物的磁性、顏色、結構和相對穩定性方面,它可以出奇地準確。
考慮一個帶正電的金屬離子,例如“場”中六個帶負電的配體(例如CN-)中的Fe3+。我們需要考慮兩個能量項。第一個是金屬和配體之間的靜電吸引,它與它們之間的距離成反比

第二個項是當第三個電子新增到填充軌道時,由泡利不相容原理產生的排斥。除了進入更高能量的反鍵軌道外,這個第三個電子無處可去。當配體孤對電子接近佔據的金屬d軌道時,就會出現這種情況


現在讓我們考慮當金屬離子與配體結合在一起時,這些吸引力和排斥項的影響。我們分兩步進行:首先在金屬周圍形成一個配體“球”,然後將六個配體移動到八面體的頂點。最初,所有五個d軌道都是簡併的,即由於對稱性,它們具有相同的能量。在第一步中,反鍵相互作用使軌道的能量升高,但它們仍然是簡併的。在第二步中,d軌道分裂成兩個對稱性類別,一個較低能量的三重簡併組(t2g軌道)和一個較高能量的二重簡併組(eg軌道)。
eg和t2g軌道之間的能量差用符號ΔO表示,其中“O”代表“八面體”。我們將看到,這種分裂能對軌道重疊的程度敏感,因此取決於金屬和配體。相對於中點能量(重心),t2g軌道被穩定了2/5 ΔO,而eg軌道在八面體配合物中被不穩定了3/5 ΔO。

是什麼導致d軌道分裂成兩組?回想一下,d軌道相對於笛卡爾座標軸具有特定的取向。dxy、dxz和dyz軌道的葉瓣(t2g軌道)分別位於xy-、xz-和yz-平面。這三個d軌道在x-、y-和z-方向上具有節點。包含配體孤對電子的軌道沿這些軸取向,因此與金屬t2g軌道沒有重疊。很容易看出,這三個d軌道必須透過對稱性是簡併的。另一方面,dz2和dx2-y2軌道的葉瓣(eg軌道)直接指向鍵合軸,並與配體軌道有很強的重疊。雖然不太直觀,但這些軌道也透過對稱性是簡併的,並且具有反鍵特性。
比較晶體場理論和分子軌道理論(在此上下文中也稱為配體場理論)對於八面體過渡金屬配合物的結果是有益的。下面的能級圖對d1八面體離子[Ti(H2O)6]3+進行了比較。在右側的MO圖中,前沿軌道來自金屬d軌道。較低的t2g組包含一個電子,由於對稱性是非鍵合的,而eg軌道是反鍵合的。金屬4s軌道具有a1g對稱性,形成一個配體中心的低能鍵合組合,以及一個金屬中心的、高於eg能級的反鍵合組合。這就是為什麼我們的d電子計數規則不需要考慮金屬4s軌道的原因。重要的收穫資訊是,對於過渡金屬配合物的前沿軌道,晶體場理論和MO理論給出了非常相似的結果。
.png)

-nitrate-photo.jpg)
強場配體和弱場配體。分光化學序列根據其八面體配合物中t2g和eg軌道之間的能量差ΔO對配體進行排序。此能量差透過這些能級之間的光譜躍遷來測量,該躍遷通常位於光譜的可見光部分,並導致具有部分填充d軌道的配合物的顏色。產生較大分裂的配體稱為強場配體,而產生較小分裂的配體稱為弱場配體。
簡化的分光化學序列為
弱場 I- < Br- < Cl- < NO3- < F- < OH- < H2O < 吡啶 < NH3 < NO2- < CN- < CO 強場

-nitrate-xtal-1973-unit-cell-CM-3D-balls.png)
軌道重疊。參考上面的分子軌道圖,我們看到d電子能級之間的分裂反映了eg金屬軌道與配體之間的反鍵相互作用。因此,我們預計配體場強度與金屬-配體軌道重疊相關。透過非常電負性的原子(例如O和鹵素)鍵合的配體預計為弱場,而透過C或P鍵合的配體通常為強場。透過N鍵合的配體強度中等。換句話說,硬鹼傾向於成為弱場配體,軟鹼是強場配體。
- 能量單位。能量可以透過多種方式計算,並且嘗試將分裂能ΔO與鍵能等更熟悉的量聯絡起來非常有用。
- 當ΔO透過光學測量時,一個波長為λ的光子被吸收,電子從t2g軌道躍遷到eg軌道。光子能量與其波長和頻率的關係為
- E = hν = hc/λ = hc

- 這裡ν是電磁輻射的頻率,h是普朗克常數(6.626x10-34 J*s),c是光速。稱為“波數”,是波長的倒數,通常以cm-1為單位。光譜學家通常用波數表示能隙。
- 例如,紅光子的波長約為620 nm,波數約為16,000 cm-1。用其他能量單位表示,相同的紅光子的能量為2.0 eV(1 eV = 1240 nm)或193 kJ/mol(1 eV = 96.5 kJ/mol)。如果我們將其與碳-碳單鍵的解離能(350 kJ/mol)進行比較,我們會發現C-C鍵的能量約為紅光子的兩倍。我們需要紫外光子(E > 350 kJ/mol = 3.6 eV = 345 nm = 29,000cm-1)才能斷裂C-C鍵。
我們將看到ΔO在過渡金屬配合物中變化很大,從近紅外到紫外波長。因此,t2g和eg軌道之間的能量差可以在相當弱的共價鍵到相當強的共價鍵的能量之間變化。
ΔO取決於金屬和配體。我們可以透過比較一系列d6金屬配合物來了解ΔO的趨勢
配合物 ΔO (cm-1) [Co(H2O)6]2+ 9,300 [Co(H2O)6]3+ 18,200 [Co(CN)6]3- 33,500 [Rh(H2O)6]3+ 27,000 [Rh(CN)6]3- 45,500
ΔO的重要趨勢:
- 具有相同配體的Co3+配合物的ΔO大於Co2+配合物。這反映了晶體場分裂的靜電性質。
- Rh3+配合物的ΔO大於Co3+配合物。一般來說,第二和第三過渡系元素(4d和5d元素)的分裂更大,而不是3d系元素。
- 對於一種氧化態的給定金屬(例如,Co3+),ΔO的趨勢遵循分光化學序列。因此,含有強場配體CN-的[Co(CN)6]3-的ΔO大於含有弱場配體H2O的[Co(H2O)6]3+的ΔO。

4d和5d元素的尺寸和化學性質相似。在比較3d、4d和5d系配合物的ΔO值時(例如,比較Co、Rh、Ir或Fe、Ru、Os三元組中的元素),我們總是發現3d << 4d ≲ 5d。這種趨勢反映了d軌道的空間範圍,以及它們與配體軌道的重疊。3d軌道較小,並且它們在成鍵方面不如4d或5d有效。由於鑭系收縮,4d和5d軌道彼此相似。在5d系的開始(56Ba和72Hf之間)是14個鑭系元素(57La - 71Lu)。
儘管5d元素的價軌道處於比4d元素更高的主量子層,但在穿過鑭系元素的過程中,原子核增加了14個質子,導致原子軌道的尺寸收縮。重要的結果是,4d和5d元素的價軌道具有相似的尺寸,因此這些元素在化學性質上的相似性遠大於它們與3d系中的同族元素的相似性。例如,Ru的化學性質與Os非常相似,如右側所示,但與Fe的化學性質大不相同。
過渡金屬配合物的顏色。觀察過渡金屬配合物的顏色是一種簡單、定性的方法來了解相對的晶體場分裂能ΔO。吸收的光子能量越高,能隙越大。然而,配合物吸收的顏色與其呈現的顏色(即它反射的光的顏色)互補,在色輪上與吸收的顏色相反。

示例:(所有d7 Co2+配合物)
[Co(H2O)6]2+在其鹽和濃溶液中呈現紫色,因為它吸收綠色範圍的光。
[Co(NH3)6]2+呈稻草色,因為它吸收藍色範圍的光。
[Co(CN)6]4-呈紅色,吸收光譜的紫外和紫外部分。這與CN-是比NH3更強的場配體的觀點一致,因為UV光子的能量高於紅橙光子的能量。
此方法適用於大多數過渡金屬配合物,因為它們中的大多數都吸收可見光範圍(400-700 nm = 25,000到14,300 cm-1)中的某個波長,或者具有延伸到可見光的紫外躍遷,使它們看起來呈黃色;然而,也有一些配合物,如[Rh(CN)6]3-,因為它們的d-d躍遷位於紫外區域,所以看起來無色。其他配合物,如[Mn(H2O)]62+,顏色較淺,因為它們的d-d躍遷涉及配合物自旋狀態的變化。

導致CO、CN-和膦等配體具有高配體場強的一個重要因素是金屬與配體之間的π鍵。金屬配合物中存在三種類型的π鍵

最常見的情況是,當像一氧化碳或氰化物這樣的配體將其σ(非鍵合)電子給予金屬,同時透過金屬t2g軌道和配體π*軌道之間的重疊從金屬接受電子密度時。這種情況稱為“反鍵”,因為配體將σ電子密度給予金屬,而金屬將π電子密度給予配體。因此,配體充當σ-給體和π-受體。在π反鍵中,金屬將π電子給予配體π*軌道,在反鍵分子軌道中增加電子密度。這導致C-O鍵減弱,這在實驗上觀察到為鍵的伸長(相對於氣相中的自由CO)和C-O紅外伸縮頻率的降低。
d-d π 鍵發生在諸如磷之類的元素與金屬鍵合時,磷具有σ對稱的孤對電子和一個空的3d軌道,而金屬則在t2g軌道中具有電子。對於與低價晚過渡金屬鍵合的膦配合物(例如,三苯基膦),這種情況很常見。在這種情況下,反鍵合類似於CO的例子,只是受體軌道是磷的3d軌道而不是配體的π*軌道。這裡,膦配體充當σ-給體和π-受體,形成dπ-dπ鍵。
第三種金屬-配體π鍵發生在π-給體配體(一種既具有σ對稱電子對又具有填充的正交p軌道的元素)與金屬鍵合時,如上圖右側所示,例如O2-配體。這發生在早期過渡金屬配合物中。在這個例子中,O2-同時充當σ-給體和π-給體。這種相互作用通常被描繪成金屬-配體多重鍵,例如,氧釩陽離子[VO]2+中的V=O鍵。典型的π-給體配體有氧化物(O2-)、氮化物(N3-)、醯胺(RN2-)、醇鹽(RO-)、醯胺(R2N-)和氟化物(F-)。對於晚過渡金屬,強π-給體與填充的d能級形成反鍵相互作用,這對自旋態、氧化還原電位和配體交換速率產生影響。π-給體配體在光譜化學序列中位置較低。[5]


含碳的π-給體配體及其與過渡金屬離子的配合物在烯烴複分解反應中非常重要,該反應中碳-碳雙鍵發生交換。使用這些催化劑,環狀烯烴可以透過開環複分解聚合(ROMP)高產率地轉化為線性聚合物。這種型別的催化劑是由Richard Schrock和Robert Grubbs的研究小組開發的,他們與Yves Chauvin共同獲得了2005年諾貝爾化學獎,以表彰他們的發現。Schrock催化劑基於早期過渡金屬如Mo;它們反應性更強,但對不同的有機官能團和質子溶劑的耐受性不如Grubbs催化劑,後者基於Ru配合物。
過渡金屬配合物中d軌道分裂成不同能級的現象對其穩定性、反應性和磁性具有重要影響。讓我們首先考慮八面體配合物[M(H2O)6]3+的簡單情況,其中M = Ti、V、Cr。由於配合物是八面體,它們都具有相同的能級圖。


Ti3+、V3+和Cr3+配合物分別具有一個、兩個和三個d電子,它們分別填充簡併的t2g軌道。根據洪特規則,自旋平行排列,洪特規則指出,最低能量狀態具有最高的自旋角動量。
對於每個配合物,我們都可以計算一個晶體場穩定化能,CFSE,它是配合物處於基態與處於所有五個d軌道都處於能量重心處的假設狀態之間的能量差。
- 對於Ti3+,有一個電子被穩定了2/5 ΔO,所以CFSE = -(1)(2/5)(ΔO) = -2/5 ΔO。
- 類似地,V3+和Cr3+的CFSE分別為-4/5 ΔO和-6/5 ΔO。
對於Cr2+配合物,它有四個d電子,情況更復雜。現在我們可以得到高自旋構型(t2g)3(eg)1,或者低自旋構型(t2g)4(eg)0,其中兩個電子配對。這兩種狀態的能量是多少?
- 高自旋:CFSE = (-3)(2/5)ΔO + (1)(3/5)ΔO = -3/5 ΔO
- 低自旋:CFSE = (-4)(2/5)ΔO + P = -8/5 ΔO + P,其中P是配對能
- 能量差 = -8/5 ΔO + P - (-3/5 ΔO) = -ΔO + P
配對能P是在同一軌道中放置兩個電子的能量損失,這是由於電子之間的靜電排斥引起的。對於3d元素,P的典型值為約15,000 cm-1。
這裡的重要結果是,如果ΔO > P,則配合物將是低自旋的,如果ΔO < P,則配合物將是高自旋的。
由於ΔO取決於金屬和配體,因此它決定了配合物的自旋態。

經驗法則
- 3d配合物在與弱場配體結合時為高自旋,在與強場配體結合時為低自旋。
- 高價3d配合物(例如,Co3+配合物)傾向於低自旋(ΔO較大)
- 4d和5d配合物始終為低自旋(ΔO較大)
請注意,高自旋和低自旋狀態僅發生在具有4到7個d電子的3d金屬配合物中。具有1到3個d電子的配合物可以在t2g組的各個軌道中容納所有電子。具有8、9或10個d電子的配合物將始終具有完全填充的t2g軌道和2-4個eg組中的電子。
高自旋和低自旋配合物的例子
- [Co(H2O)62+]包含一個d7金屬離子,以及一個弱場配體。根據磁化率測量,已知該配合物為高自旋,每個分子具有三個未成對電子。其軌道佔據情況為(t2g)5(eg)2。
- 我們可以計算CFSE為-(5)(2/5)ΔO + (2)(3/5)ΔO = -4/5 ΔO。
- [Co(CN)64-]也是一個八面體d7配合物,但它包含CN-,這是一種強場配體。其軌道佔據情況為(t2g)6(eg)1,因此它有一個未成對電子。
- 在這種情況下,CFSE為-(6)(2/5)ΔO + (1)(3/5)ΔO + P = -9/5 ΔO + P。
過渡金屬配合物的磁性
具有未成對電子的化合物具有固有的磁矩,該磁矩源於電子自旋。此類化合物與外加磁場發生強烈的相互作用。它們的磁化率提供了一種簡單的方法來測量過渡金屬配合物中未成對電子的數量。
如果過渡金屬配合物沒有未成對電子,則它是抗磁性的,並且會被弱排斥到不均勻磁場的高場區域。具有未成對電子的配合物通常是順磁性的。順磁體中的自旋在施加的磁場中獨立排列,但在沒有磁場的情況下不會自發排列。此類化合物會被磁鐵吸引,即它們會被拉入不均勻磁場的高場區域。吸引力可以用古伊天平或超導量子干涉儀磁強計測量,它與配合物的磁化率(χ)成正比。
在沒有自旋-軌道耦合的情況下,離子的有效磁矩(µeff)由其自旋和軌道矩之和給出
- µeff = µspin + µorbital = µs + µL
在八面體3d金屬配合物中,軌道角動量在很大程度上被對稱性“猝滅”,因此我們可以近似為
- µeff ≈ µs
我們可以使用以下公式根據未成對電子數(n)計算µs

這裡µB是玻爾磁子(= eh/4πme)= 9.3 x 10-24 J/T。這個僅自旋公式對於第一排過渡金屬配合物來說是一個很好的近似,尤其是高自旋配合物。下表比較了具有1-5個未成對電子的八面體配合物的µeff的計算值和實驗測量值。
離子 未成對
電子
數目僅自旋
磁矩 /μB觀察值
磁矩 /μBTi3+ 1 1.73 1.73 V4+ 1 1.68–1.78 Cu2+ 1 1.70–2.20 V3+ 2 2.83 2.75–2.85 Ni2+ 2 2.8–3.5 V2+ 3 3.87 3.80–3.90 Cr3+ 3 3.70–3.90 Co2+ 3 4.3–5.0 Mn4+ 3 3.80–4.0 Cr2+ 4 4.90 4.75–4.90 Fe2+ 4 5.1–5.7 Mn2+ 5 5.92 5.65–6.10 Fe3+ 5 5.7–6.0
這些八面體配合物與僅自旋公式存在的小偏差可能是由於忽略了軌道角動量或自旋-軌道耦合造成的。四面體d3、d4、d8和d9配合物往往比相同離子的八面體配合物表現出更大的偏離僅自旋公式的現象,因為在四面體情況下,軌道貢獻的猝滅效果較差。
高自旋和低自旋配合物規則總結

- 3d配合物:可以是高自旋或低自旋,具體取決於配體(d4、d5、d6、d7)
- 4d和5d配合物:始終為低自旋,因為ΔO較大
- 最大CFSE出現在d3和d8情況下(例如,Cr3+、Ni2+),與弱場配體(H2O、O2-、F-等)結合,以及在d3-d6與強場配體結合時(Fe2+、Ru2+、Os2+、Co3+、Rh3+、Ir3+等)。
- Irving-Williams 級數。對於 M2+ 配合物,配合物的穩定性遵循以下順序:Mg2+ < Mn2+ < Fe2+ < Co2+ < Ni2+ < Cu2+ > Zn2+。這種趨勢表示隨著離子變小(在元素週期表中從左到右),路易斯酸性增加,同時也反映了 CFSE 的趨勢。氣相 M2+ 離子的水合焓也反映了同樣的趨勢,如右側圖表所示。請注意,Ca2+、Mn2+ 和 Zn2+ 分別是 d0、d5(高自旋)和 d10 水合離子,它們都具有零 CFSE,並位於同一條線上。與該線偏差最大的離子,例如 Ni2+(八面體 d8),具有最高的 CFSE。

過渡金屬配合物的顏色和光譜
過渡金屬配合物通常具有美麗的顏色,因為如上所述,它們的 d-d躍遷能量可能位於光譜的可見光部分。對於八面體配合物,這些顏色較淡(躍遷較弱),因為它們違反了拉波特選擇定則。根據該定則,在中心對稱配合物中,g → g 和 u → u 躍遷是被禁止的。d 軌道具有 g( gerade )對稱性,因此 d-d 躍遷是拉波特禁阻的。然而,當分子振動時,八面體配合物可以暫時偏離中心對稱性而吸收光。自旋翻轉也受到自旋選擇定則的光學躍遷禁阻,因此激發態將始終與基態具有相同自旋多重度。
即使是最簡單的過渡金屬配合物的光譜也相當複雜,因為 d 電子填充 t2g 和 eg 軌道的多種可能方式。例如,如果我們考慮一個 d2 配合物,例如 V3+(aq),我們知道這兩個電子可以位於五個 d 軌道中的任何一個,並且可以是自旋向上或自旋向下。對於 d2 配合物,實際上有 45 種這樣的排列(稱為微觀態),它們不違反泡利不相容原理。通常我們只關心能量最低的六個,其中兩個電子佔據 t2g 集中各個軌道,並且它們的自旋全部向上或向下對齊。
3%2B.png)
當我們考慮 [Cr(NH3)6]3+ 離子的 d-d 躍遷時,我們可以看到這些微觀態如何在電子光譜中發揮作用。該離子為 d3,因此三個 t2g 軌道中的每一個都包含一個未成對電子。我們預計當 t2g 軌道中的三個電子之一被激發到空的 eg 軌道時會發生躍遷。有趣的是,我們發現可見光區不僅有一個,而是兩個躍遷。
我們看到兩個躍遷的原因是電子可以來自 t2g 軌道中的任何一個,並最終進入 eg 軌道中的任何一個。為了論證起見,讓我們假設電子最初位於 dxy 軌道中。它可以被激發到 dz2 或 dx2-y2 軌道
- dxy --> dz2 (較高能量)
- dxy --> dx2-y2 (較低能量)
第一個躍遷的能量較高(波長較短),因為在激發態時,構型為 (dyz1dxz1dz21)。所有三個激發態軌道都具有一定的 z 分量,因此 d 電子密度沿 z 軸“堆積”。因此,電子-電子排斥增加了該躍遷的能量。在第二種情況下,激發態構型為 (dyz1dxz1dx2-y21),並且 d 電子更對稱地分佈在金屬周圍。這種效應導致 d-d 帶分裂約 8,000 cm-1。我們可以透過對稱性證明所有其他可能的躍遷都等效於這兩個躍遷之一,因此我們只看到 Cr3+ 配合物的兩個可見吸收帶。


過渡金屬配合物最重要的非八面體幾何形狀是
- 4 配位:平面正方形和四面體
- 5 配位:四方錐和三角雙錐


非八面體幾何形狀中 d 軌道的能量。左側的圖顯示了當我們透過沿 z 軸拉長八面體配合物(四方畸變)、去除一個配體形成四方錐或去除沿 z 軸的兩個配體形成平面正方形配合物時,d 軌道能級圖會發生什麼變化。在所有情況下,我們透過使 xy 平面中的鍵變短來保持總鍵級相同,因為 z 方向的鍵被拉伸和/或斷裂。
偏離八面體對稱性的畸變打破了 t2g 和 eg 軌道的簡併性。具有 z 軸分量的 d 軌道(dxz、dyz、dz2)能量降低,而位於 xy 平面中的軌道(dxy、dx2-y2)能量升高。重心(加權平均軌道能量)保持不變。此外,需要注意的是,dxy 和 dx2-y2 軌道之間的分裂在 ΔO 上是恆定的,而不管畸變的性質如何。在平面正方形幾何形狀中,dxz、dyz、dz2、dxy 和 dx2-y2 軌道的能量分別為 -0.51、-0.40、+0.21 和 +1.21(以 ΔO 為單位)。
為什麼“快樂”的八面體配合物會想要失去兩個配體形成平面正方形配合物?這在 d8 和有時在 d9 配合物中經常發生,並且具有較大的 ΔO,即具有強場配體的 3d8 配合物以及具有任何配體的 4d8、5d8 配合物。這種 d8 配合物的例子包括 [Ni(CN)4]2-、抗癌藥物順鉑(順式-Pt(NH3)2Cl2)、[Pd(H2O)4]2+ 和 [AuCl4]-。在 d8 電子數下,最低四個軌道被填充,最高軌道(dx2-y2)為空,導致較大的 CFSE(2.4 ΔO,而八面體 d8 為 1.2 ΔO)。對於 4d8 和 5d8 配合物以及具有強場配體的 3d8 配合物,這 1.2 ΔO 的差異超過了配對能量。這些平面正方形配合物是反磁性的,並且往往非常穩定。對於弱場配體,3d8 配合物是八面體和順磁性的(例如 [Ni(H2O)6]2+,其在 eg 軌道中具有兩個未成對電子)。
催化中的平面正方形配合物

平面正方形 d8 配合物可以透過兩個電子被氧化成八面體(低自旋)d6 配合物,後者也具有較大的 CFSE。由於失去兩個電子伴隨著獲得兩個配體,因此該過程稱為氧化加成。相反的過程稱為還原消除。這兩個過程一起在催化迴圈中發揮作用,例如使用威爾金森催化劑進行烯烴的氫化。[10][11]左側顯示了催化迴圈。
催化劑在4配位的Rh(I) (4d8)和6配位的Rh(III) (4d6)之間迴圈。該絡合物首先氧化加成H2,生成一個六配位絡合物,其中氫的形式為H-。烯烴分子利用其π電子與金屬配位,取代溶劑分子。該絡合物透過將烯烴插入金屬-氫鍵中進行重排,這一過程稱為**遷移插入**。最後,該絡合物透過消除氫化烯烴(還原消除)恢復到平面正方形幾何構型。威爾金森催化劑活性很高,廣泛用於均相氫化、硼氫化和氫矽烷化反應。[12][13]使用手性膦配體,該催化劑可以將前手性烯烴氫化得到對映體純產物。[14]使用佔據平面正方形絡合物四個配位位點中的三個的五配位手性配體,可以生產非常高產率的對映體純氫化產物。類似的手性Ir(I)絡合物催化前手性酮的氫化生成手性伯醇,這是許多手性藥物化合物生產中的重要步驟。[15]
-3D-vdW.png)
線性ML2絡合物。Cu(I)、Ag(I)和Au(I)離子與弱場和強場配體都形成線性ML2絡合物。例如,在氰化物鹽存在下,金或銀金屬發生空氣氧化,形成[Ag(CN)2]-或[Au(CN)2]-,這種氧化還原反應被用於開採這些貴金屬。不溶性的Ag(I)化合物,例如AgCl,可以在氨溶液中溶解形成可溶性線性絡合物,例如[Ag(NH3)2]+。
線性配位幾何結構源於s和d軌道的雜化。例如,在上圖所示的[Au(CN)2]-離子中,5dz2和6s軌道的雜化體各含有一個電子,並沿z軸方向,類似於HC≡CH等分子中pz和s軌道雜化的方式。在這些線性絡合物中,晶體場分裂成三個能級,其中填充的dxy和dx2-y2軌道能量最低,填充的dxz和dyz軌道能量居中,半填充的dz2軌道能量最高。dxz、dyz軌道與CN- π*軌道之間的反饋鍵也發生了,進一步穩定了絡合物。[16][17]
5.7 Jahn-Teller效應
[edit | edit source]
Jahn–Teller效應,有時也稱為Jahn–Teller畸變,描述了與某些電子構型相關的分子和離子的幾何畸變。這種電子效應以赫爾曼·阿瑟·雅恩和愛德華·泰勒的名字命名,他們使用群論證明了軌道簡併的分子不可能是穩定的。[18]Jahn–Teller定理本質上指出,任何具有空間簡併電子基態的非線性分子都會發生幾何畸變,從而消除這種簡併性,因為畸變降低了分子的總能量。
我們可以在八面體金屬絡合物的背景下理解這種效應,方法是考慮eg軌道集中包含一個或三個電子的d電子構型。其中最常見的是高自旋d4(例如,CrF2)、低自旋d7(例如,NaNiO2)和d9(例如,Cu2+)。如果絡合物可以發生畸變以破壞對稱性,則一個(以前)簡併的eg軌道能量將降低,另一個能量將升高。更多的電子將佔據較低的軌道而不是較高的軌道,導致電子能量的整體降低。當t2軌道部分填充時,四面體絡合物中也會發生類似的畸變。這種降低電子能量的幾何畸變被稱為電子驅動的。類似的電子驅動的畸變發生在一維鏈狀化合物中,在那裡它們被稱為Peierls畸變,以及二維鍵合薄片中,在那裡它們被稱為電荷密度波。
-3D-balls.png)

Jahn–Teller效應最常出現在八面體絡合物中,尤其是六配位銅(II)絡合物。[19]該離子的d9電子構型在兩個簡併的eg軌道中產生了三個電子,導致雙重簡併的電子基態。這種絡合物沿分子四重軸之一(始終標記為z軸)發生畸變,這會消除軌道和電子簡併性並降低總能量。畸變通常採用沿z軸延長與配體鍵合的方式,但偶爾也表現為這些鍵的縮短(Jahn–Teller定理並沒有預測畸變的方向,只預測了不穩定幾何構型的存在)。當發生這種伸長時,其作用是降低路易斯鹼性配體上的電子對與任何具有z分量的軌道中的電子之間的靜電排斥,從而降低絡合物的能量。如果未畸變的絡合物預計具有反演中心,則在畸變後將保持該中心。
在八面體絡合物中,當奇數個電子佔據eg軌道時,Jahn–Teller效應最為明顯。這種情況出現在具有d9、低自旋d7或高自旋d4構型的絡合物中,所有這些絡合物都具有雙重簡併的基態。在這些化合物中,參與簡併的eg軌道直接指向配體,因此畸變會導致較大的能量穩定。嚴格來說,當由於t2g軌道中的電子而導致簡併時,也會發生這種效應(即諸如d1或d2之類的構型,它們都是三重簡併的)。然而,在這種情況下,這種效應不太明顯,因為在使配體遠離t2g軌道的過程中,排斥的降低要小得多,而t2g軌道並沒有直接指向配體(見下表)。四面體絡合物(例如錳酸鹽([MnO4]2-,d1)也是如此:畸變非常細微,因為當配體沒有直接指向軌道時,獲得的穩定性較小。
八面體配位的預期效應在下表中給出。
| d電子數 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 高/低自旋 | 高自旋 | 低自旋 | 高自旋 | 低自旋 | 高自旋 | 低自旋 | 高自旋 | 低自旋 | ||||||
| J-T效應強度 | 弱 | 弱 | 強 | 弱 | 弱 | 弱 | 弱 | 強 | 強 | |||||
弱:弱Jahn–Teller效應(t2g軌道不均勻佔據)
強:預期強Jahn–Teller效應(eg軌道不均勻佔據)
空白:預期無Jahn–Teller效應。
Jahn–Teller效應在某些化合物的紫外-可見吸收光譜中表現出來,在那裡它通常會導致譜帶分裂。它在許多銅(II)絡合物的結構中很容易顯現。[20]可以從低溫電子自旋共振光譜的精細結構獲得有關此類絡合物各向異性和配體結合性質的更多詳細資訊。
5.8 四面體絡合物
[edit | edit source]四面體絡合物由後過渡金屬離子(Co2+、Cu2+、Zn2+、Cd2+)和一些早期過渡金屬(Ti4+、Mn2+)形成,尤其是在配體較大的情況下。在這些情況下,小的金屬離子不容易容納大於4的配位數。四面體離子和小分子的例子有[CoCl4]2-、[MnCl4]2-和TiX4(X = 鹵素)。在一些氧陰離子中也觀察到四面體配位,例如[FeO4]4-,它作為離散陰離子存在於鹽Na4FeO4和Sr2FeO4中,以及在中性氧化物RuO4和OsO4中。金屬羰基絡合物Ni(CO)4和Co(CO)4]-也是四面體。


可以透過連線四面體的頂點形成一個立方體來理解四面體晶體場中d軌道的分裂,如左側的圖片所示。四面體M-L鍵位於立方體的體對角線上。dz2和dx2-y2軌道指向笛卡爾座標軸,即指向立方體的面,並且與配體孤對電子的接觸最少。因此,這兩個軌道形成一個低能量的雙重簡併e組。dxy、dyz和dxz軌道指向立方體的邊緣,並形成一個三重簡併t2組。雖然t2軌道與配體軌道的重疊比e組多,但與八面體絡合物的eg軌道相比,它們仍然是弱相互作用的。
所得晶體場能級圖如圖右側所示。分裂能Δt大約是使用相同配體形成的八面體絡合物分裂能的4/9。對於3d元素,Δt因此相對於配對能很小,並且它們的四面體絡合物總是高自旋。請注意,我們已經刪除了“g”下標,因為四面體沒有對稱中心。
四面體配合物通常具有鮮豔的顏色,因為它們缺乏對稱中心,而對稱中心會禁止 d-d*躍遷。由於低能級躍遷是允許的,因此這些配合物通常在可見光範圍內吸收,並且消光係數比相應的八面體配合物高 1-2 個數量級。在乾燥劑中可以看到這種效應的一個例子,乾燥劑包含無色無水硫酸鈣(石膏)顆粒,這些顆粒可以吸收氣體中的水分。乾燥劑中的指示劑染料是氯化鈷(II),溼潤時為淺粉色(八面體),乾燥時為深藍色(四面體)。可逆水合反應為
- Co[CoCl4] + 12 H2O ⇌ 2 [Co(H2O)6]Cl2
- (深藍色,四面體 [CoCl4]2-) (淺粉色,八面體 [Co(H2O)6]2+)

乾燥劑乾溼狀態 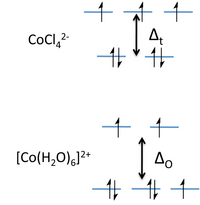


晶體場穩定化能 (CFSE) 是過渡金屬配合物穩定性的一個重要因素。具有高 CFSE 的配合物傾向於熱力學穩定(即,它們具有較高的 Ka 值,Ka 是金屬-配體締合的平衡常數),並且也動力學惰性。它們在動力學上是惰性的,因為配體取代需要它們在過渡態中解離(失去一個配體),締合(獲得一個配體)或交換(同時獲得和失去配體)。如果 CFSE 很大,即使配體交換反應的產物也是穩定的配合物,這些配位幾何的扭曲也會導致較大的活化能。因此,Pt4+、Ir3+(均為低自旋 5d6)和 Pt2+(平面正方形 5d8)的配合物具有非常緩慢的配體交換速率。
還有其他兩個重要因素有助於配合物的穩定性
- 金屬和配體的硬軟相互作用(與配合物形成的能量有關)
- 螯合效應,它是配合物穩定性的熵貢獻者。
硬軟相互作用
硬酸通常是小尺寸、高電荷密度、弱極化性陽離子,例如 H+、Li+、Na+、Be2+、Mg2+、Al3+、Ti4+ 和 Cr6+。電正性金屬處於高氧化態通常是硬酸。這些元素主要存在於氧化物礦物中,因為 O2- 是硬鹼。
一些硬鹼包括 H2O、OH-、O2-、F-、NO3-、Cl- 和 NH3。
硬酸-鹼相互作用主要是靜電的。硬酸與硬鹼的配合物由於 CFSE 的靜電成分而穩定。
軟酸是大尺寸、可極化、電負性金屬離子處於低氧化態,例如 Ni0、Hg2+、Cd2+、Cu+、Ag+ 和 Au+。
軟鹼是陰離子/中性鹼,例如 H-、C2H4、CO、PR3、R2S 和 CN-)。軟酸通常以硫化物或砷化物礦物形式存在於自然界中。
軟酸和軟鹼之間的鍵合主要是共價的。例如,金屬羰基透過零價或低價金屬與中性 CO 之間的共價相互作用結合形成 Ni(CO)4、Fe(CO)5、Co(CO)4-、Mn2(CO)10、W(CO)6 及相關化合物。
硬硬和軟軟相互作用的偏好(“相似相吸”)在鹵化銅的性質中得到了很好的說明
- CuF CuI
- 不穩定 穩定
- CuF2 CuI2
- 穩定 不穩定
CuF 和 CuI2 化合物從未被分離出來,並且在熱力學上不穩定,會發生歧化反應
- 2 CuF(s) → Cu(s) + CuF2(s)
- 2 CuI2(s) → 2 CuI(s) + I2(s)
我們將在第 9 章中進一步瞭解這些化合物的能量學定量。

含有兩個以上金屬離子結合位點的配體稱為螯合配體(源於希臘語 χηλή,chēlē,意為“爪”)。顧名思義,螯合配體對金屬離子的親和力高於僅具有一個結合基團(稱為單齒 =“單齒”)的配體。
考慮水溶液中的兩個絡合平衡,一方面是鈷(II)離子 Co2+(aq) 與乙二胺 (en),另一方面是氨 NH3。
- [Co(H2O)6]2+ + 6 NH3 ⇌ [Co(NH3)6]2+ + 6 H2O (1)
- [Co(H2O)6]2+ + 3 en ⇌ [Co(en)3]2+ + 6 H2O (2)
在電子方面,氨和 en 配體非常相似,因為兩者都透過 N 結合,並且它們的氮原子的路易斯鹼強度相似。這意味著兩個反應的 ΔH° 必須非常相似,因為在每種情況下都會形成六個 Co-N 鍵。然而,有趣的是,我們觀察到第二個反應的平衡常數比第一個反應大 100,000 倍。
這兩個反應之間的主要區別在於,第二個反應涉及更少粒子的“縮合”來形成配合物。這意味著這兩個反應的熵變是不同的。第一個反應的 ΔS° 值接近於零,因為方程兩側的分子數相同。第二個反應的 ΔS° 為正,因為四個分子聚合在一起,但產生了七個分子。它們之間的差異 (ΔΔS°) 約為 +100 J/mol-K。我們可以使用以下公式將其轉換為平衡常數的比率
- Kf(en)/Kf(NH3) = e-ΔΔG°/RT ≈ e+ΔΔS°/R ≈ e12 ≈ 105


底線是螯合效應是熵驅動的。由此可見,配體包含的結合基團越多,ΔS° 越正,配合物形成的 Kf 就越高。在這方面,六齒配體乙二胺四乙酸 (EDTA) 是形成八面體配合物的最佳配體,因為它具有六個結合基團。在所有四個 COOH 基團都去質子的鹼性溶液中,EDTA4- 配體的螯合效應大約為 1015。這意味著,對於給定的金屬離子,Kf 對於 EDTA4- 比在相同濃度下相關單齒配體的 Kf 大 1015 倍。EDTA4-牢固地結合元素週期表中幾乎所有 2+、3+ 或 4+ 離子,並且是分析應用和分離的非常有用的配體。
大環效應遵循與螯合效應相同的原理,但該效應因配體的環狀構象而進一步增強。大環配體不僅是多齒的,而且由於它們以共價方式限制在其環狀形式,因此它們允許更少的構象自由度。據說配體“預組織”用於結合,並且圍繞金屬離子包裹幾乎沒有熵損失。例如,血紅素 b 是一種四齒環狀配體,它強烈絡合過渡金屬離子,包括(在生物系統中)Fe+2。
下面顯示了一些其他常見的螯合和環狀配體


乙醯丙酮 (acac-,右) 是一種陰離子雙齒配體,透過兩個氧原子與金屬離子配位。acac- 是硬鹼,因此它更喜歡硬酸陽離子。對於二價金屬離子,acac- 形成中性揮發性配合物,例如 Cu(acac)2 和 Mo(acac)2,這些配合物可用於金屬薄膜的化學氣相沉積 (CVD)。
2,2'-聯吡啶和相關的雙齒配體,如 1,10-菲咯啉(下方,左中)與 Ru2+ 等金屬形成螺旋槳狀配合物。[Ru(bpy)3]2+ 配合物(下方左)具有光致發光性,也可以發生光氧化還原反應,使其成為光催化和人工光合作用的有趣化合物。[Ru(bpy)3]2+ 等金屬多吡啶配合物的螺旋槳形狀巧合地與 DNA 主溝的大小和螺旋性相匹配。這導致了對沿著 DNA 骨架進行電子轉移反應的一些有趣的研究,這些反應由金屬配合物的光激發引發。
冠醚,如 18-冠-6(下方,右中),是環狀硬鹼,可以絡合鹼金屬陽離子。根據環中環氧乙烷單元的數量,冠醚可以選擇性地結合 Li+、Na+ 或 K+。
- 冠醚的螯合性質與天然抗生素纈氨黴素(右下圖)類似,後者可以選擇性地將K+離子跨過細菌細胞膜運輸,透過耗散其膜電位殺死細菌。與冠醚一樣,纈氨黴素也是一種環狀硬鹼。




過渡金屬配合物可以交換一個配體以換取另一個配體,這些反應在其合成、立體化學和催化化學中都很重要。化學反應的機理與其反應動力學密切相關。與有機化學一樣,過渡金屬反應的機理通常是從實驗中推斷出來的,這些實驗檢查了進入和離開配體對反應速率的濃度依賴性、中間體的檢測以及反應物和產物的立體化學。
熱力學與動力學。當我們考慮過渡金屬配合物的反應時,務必記住其熱力學和動力學之間的區別。例如,形成平面四方形四氰合鎳酸鹽配合物
- Ni2+(aq) + 4 CN-(aq) ⇌ [Ni(CN)4]2- (aq) Keq ≈ 1030 M-4
從熱力學上講,[Ni(CN)4]2-非常穩定,這意味著上述平衡非常偏向於右側。然而,從動力學上講,該配合物是不穩定的,這意味著它可以快速交換其配體。例如,13C標記的CN-離子與結合的CN-配體之間的交換髮生在數十毫秒的時間尺度上
- [Ni(CN)4]2- (aq) + *CN-(aq) ⇌ [Ni(CN)3(*CN)]2- + CN-(aq) kexchange ≈ 102 M-1s-1
相反,化合物可以是熱力學不穩定但動力學惰性的,這意味著它需要相對較長的時間才能發生反應。例如,[Co(NH3)6]3+離子在酸性條件下不穩定,但它與濃鹽酸的水解反應在大約一週內才能在室溫下完成
.jpg)
- [Co(NH3)6]3+(aq) + 6 H3O+(aq) ⇌ [Co(H2O)6]3+(aq) + 6 NH4+(aq) Keq ≈ 1030
亨利·陶伯透過簡單的試管實驗研究了配體交換反應的機理,將過渡金屬配合物分類為不穩定的,如果其反應半衰期為一分鐘或更短,則為惰性的,如果它們需要更長的時間才能反應。配體取代速率的動態範圍非常大,至少跨越了15個數量級。在大多數實驗室實驗的時間尺度上,陶伯對不穩定性的定義對於將反應分類為具有低活化能和高活化能的反應是有用的。正如我們將看到的,晶體場穩定化能 (CFSE)在決定活化能以及配體取代速率方面起著關鍵作用。
晶體場穩定化能和配體交換速率。讓我們考慮一個非常常見且簡單的配體交換反應,即在八面體[M(H2O)6]n+配合物中用另一個水分子取代一個水分子。由於產物(除了標記外)與反應物相同,因此我們知道此反應的ΔG° = 0且Keq = 1。透過使用同位素標記的水(通常含有17O或18O)可以透過核磁共振監測反應的程序

關於這個(否則很無聊的)反應最引人注目的事情是不同金屬離子和氧化態的速率常數的巨大差異——大約14個數量級
| Mn+ | log k (sec-1) |
|---|---|
| Cr3+ | |
| V2+ | |
| Cr2+ | |
| Cu2+ |

雖然起初似乎很奇怪,同一個離子在兩種不同的氧化態(Cr3+與Cr2+)下分別是不穩定的或不穩定的,但我們可以透過繪製配合物的d軌道分裂圖來開始解釋這種差異。我們發現,具有高CFSE(Cr3+,V2+)的八面體配合物傾向於惰性。相反,在高能eg軌道上具有電子的離子(Cr2+,Cu2+)傾向於不穩定。在Cr3+和V2+的情況下,使配合物偏離八面體對稱性(例如,形成5或7配位中間體)的能量損失特別高。對於Cr2+和Cu2+,配體取代的活化能較低,它們已經在反鍵eg軌道上具有電子。
根據我們為計算過渡金屬配合物的CFSE制定的規則,我們現在可以預測配體取代速率的趨勢
- 具有d3和d6(低自旋)構型的八面體配合物,例如Cr3+(d3)、Co3+(d6)、Rh3+(d6)、Ru2+(d6)和Os2+(d6)由於其高CFSE而傾向於取代惰性。
- 平面正方形d8配合物,尤其是4d和5d系列中的配合物,也取代惰性。例如,Pd2+、Pt2+和Au3+的配合物。
- 中間情況是Fe3+、V3+、V2+、Ni2+和主族離子(Be2+、Al3+)的配合物,它們是硬路易斯酸。這些配合物形成強的金屬-氧鍵,並且水交換速率在101-106 s-1範圍內。
- CFSE為零的離子在納秒的時間尺度上交換水分子(k ≈ 108-109 s-1)。這些包括具有d0、d5(高自旋)和d10電子數的離子,包括鹼金屬(Li+、Na+、K+、Rb+、Cs+)和鹼土金屬(Mg2+、Ca2+、Sr2+、Ba2+)陽離子、Zn2+、Cd2+、Hg2+和Mn2+。在這些情況下,CFSE為零,並且破壞八面體對稱性的能量成本相對較低。
- 對於p區元素,較大的離子交換速度更快(例如,Ba2+ > Ca2+和Ga3+ > Al3+),因為路易斯酸強度隨著離子尺寸的增加而降低。
- Cu2+離子(d9)作為Jahn-Teller離子,已經偏離了八面體對稱性,因此非常不穩定,以大約108 s-1的速率交換水配體。
配體取代機理。對於經歷配體取代的MLn配合物,基本上有三種不同的反應機理
- 在解離機理中,MLn配合物首先失去一個配體形成MLn-1中間體,然後進入的配體Y與MLn-1片段反應
- L(n-1)M-L* ⇌ L(n-1)M- + L* ⇌ L(n-1)M-Y
下圖說明了八面體ML6配合物的配體取代。本例中的中間態涉及三角雙錐ML5片段以及遊離的L和Y配體。

如果速率決定步驟是L從配合物中解離,則Y的濃度不影響反應速率,導致一級速率定律
![{\displaystyle {\ce {Rate={{\mathit {k}}_{1}[ML_{\mathit {n}}]}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/6f19546a5039b36de5f4c4fd594797bc0df9b999)
在八面體配合物的情況下,此反應將是一級ML6和零級Y,但前提是最高能量過渡態是ML5中間體形成之前的過渡態。如果兩個過渡態的能量接近(如右側動畫所示),則速率定律變得更加複雜。在這種情況下,我們可以透過假設MLn中間體的穩態濃度較低來簡化問題。得到的速率定律為
![{\displaystyle {\ce {Rate}}={\frac {k_{1}k_{2}[{\ce {Y}}][{\ce {ML_{\mathit {n}}}}]}{{k_{-1}[{\ce {L}}]}+k_{2}[{\ce {Y}}]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ccc9e0dfadba0e61ce35c386afdc89e901198c08)
當 k2[Y] >> k-1[L] 時,該反應簡化為更簡單的二級反應速率方程。由於過渡態的形成涉及配體的解離,因此在解離機理中,活化熵總是正的。
- 在締合機理中,進入的配體 Y 進攻 MLn 配合物,瞬時形成 MLnY 中間體,然後中間體失去一個配體 L,形成 MLn-1Y 產物。
經歷締合取代的配合物通常要麼是配位不飽和的,要麼包含一個可以改變其與金屬鍵合方式的配體,例如,硝醯配體 (NO) 的配位數或彎曲的變化。在均相催化中,締合途徑是理想的,因為結合事件,以及因此反應的選擇性,不僅取決於金屬催化劑的性質,還取決於參與催化迴圈的分子。

締合機理的例子通常存在於 d8 平方平面金屬配合物的化學中,例如 Vaska 配合物 (IrCl(CO)[P(C6H5)3]2) 和四氯鉑酸鹽(II)。這些化合物 (ML4) 與進入的 (取代的) 配體 Y 結合形成五配位中間體 ML4Y,然後在後續步驟中解離其一個配體。雖然進入的配體最初結合在赤道位置,但Berry 擬旋轉 為所有配體提供了在赤道和軸向位置之間進行取樣的低能途徑。根據微觀可逆原理,配體的解離必須發生在赤道位置。Y 的解離不會導致反應,但 L 的解離會導致淨取代,生成 d8 配合物 ML3Y。第一步通常是速率決定步驟。因此,活化熵為負,這表明過渡態的秩序增加。締合反應遵循二級動力學:產物出現速率取決於 ML4 和 Y 的濃度。

反式效應與締合機理有關,它控制著某些配體取代反應的立體化學。
反式效應是指使反式於某些其他配體的配體(後者被稱為反式導向配體)更容易發生反應。反式配體的活化歸因於電子效應,在平面正方形配合物中最為顯著,但也可在八面體配合物中觀察到。[21]順式效應 最常在八面體配合物中觀察到。
除了動力學反式效應外,反式配體也會影響分子的基態,其中最顯著的是鍵長和穩定性。一些作者更傾向於使用反式影響一詞來區分它與動力學效應,[22]而另一些作者則使用更具體的術語,如結構反式效應或熱力學反式效應。[21]
反式效應的發現歸功於Ilya Ilich Chernyaev,[23]他在 1926 年認識到它併為其命名。[24]
反式效應的強度(以反式配體取代速率的增加來衡量)遵循以下順序
- F−,H2O,OH− < NH3 < py < Cl− < Br− < I−,SCN−,NO2−,SC(NH2)2,Ph− < SO32− < PR3,AsR3,SR2,CH3− < H−,NO,CO,CN−,C2H4
請注意,弱場配體往往是較差的反式導向配體,而強場配體則是強反式導向配體。
反式效應的經典例子是順鉑及其反式異構體的合成。[25]從 PtCl42− 開始,第一個 NH3 配體隨機新增到四個等效位置中的任何一個。然而,由於 Cl− 的反式效應大於 NH3,因此第二個 NH3 新增到 Cl− 的反式位置,因此與第一個 NH3 為順式。
.svg)
另一方面,如果從 Pt(NH3)42+ 開始,則獲得反式產物。
.svg)
平方配合物中的反式效應可以用上面描述的締合機理來解釋,該機理透過三稜柱形中間體進行。具有高動力學反式效應的配體通常是那些具有高 π 酸性(如膦的情況)或低配體孤對電子-dπ 排斥(如氫化物的情況)的配體,它們更傾向於中間體中更具 π 鹼性的赤道位置。第二個赤道位置被進入的配體佔據。第三個也是最後一個赤道位置被離開的反式配體佔據,因此淨結果是動力學上有利的產物是反式於具有最大反式效應的配體的配體被消除的產物。[22]
- 交換機理類似於締合和解離途徑,只是沒有形成明顯的 MLnY 或 MLn-1 中間體。這種協同機理可以認為類似於有機化學中四面體碳原子上的 SN2 途徑的親核取代。交換機理進一步分類為締合 (Ia) 或解離 (Id),具體取決於過渡態中 M-Y 和 M-L 鍵合的相對重要性。如果過渡態的特徵是形成強的 M-Y 鍵,則機理為Ia。相反,如果 M-L 鍵的削弱在達到過渡態中更為重要,則機理為Id。
Ia 機理的一個例子是 [V(H2O)6]2+ 中本體水和配位水的交換。相反,稍緊湊的離子 [Ni(H2O)6]2+ 離子透過Id 機理交換水。[26]
離子對的影響。帶高電荷的陽離子配合物傾向於與陰離子配體形成離子對,而這些離子對通常透過Ia途徑發生反應。靜電結合的親核進入配體可以與第一配位層中的配體交換位置,從而導致淨取代。一個說明性的過程是鉻(III) 六水配合物的“加陰離子”(與陰離子反應)
- [Cr(H2O)6]3+ + SCN− ⇌ {[Cr(H2O)6], NCS}2+
- {[Cr(H2O)6], NCS}2+ ⇌ [Cr(H2O)5NCS]2+ + H2O
- 討論螯合配體及其作用,並舉一些新的例子。
- 解釋(並舉一些新的例子)我們如何知道金屬離子的八面體配合物是高自旋還是低自旋,以及我們可以進行哪些測量來確認它。
1. 預測下列配合物的分子幾何形狀,並確定每個配合物是抗磁性還是順磁性。
(a) [Fe(CN)6]3-
(b) [Ru(ox)3]4- (ox = 草酸根,C2O4)
(c) [Ag(CN)2]-
(d) [W(CO)6]
(e) [Ir(NH3)4]+
2. 對於下列每個過渡金屬配合物,給出 (i) d 電子數,(ii) 配合物的近似分子幾何形狀,以及 (iii) 顯示 d 軌道的分裂和填充的能級圖。
(a)[Os(CN)6]3-
(b)順式-PtCl2(NH3)2
(c) [Cu(NH3)4]+
3. 八面體過渡金屬配合物可以是高自旋或低自旋。四面體和平面正方形配合物是否也如此?解釋原因。
4. 對於下表中的每個過渡金屬配合物,給出與觀察到的磁矩一致的電子構型(在 t2g 和 eg 組的 3d 軌道內)。
| 化合物 | µ (BM) |
|---|---|
| [Fe(CN)6]3- | 1.8 |
| [Fe(NH3)5(H2O)]3+ | 6.1 |
| [Fe(NCS)6]4- | 5.0 |
| [Cr(acac)3] | 3.9 |
5. 對於以下每對配合物,確定晶體場穩定化能較高的配合物(並展示你的計算過程)。
(a) [Mn(CN)6]3- 與 [Mn(CN)6]4-
(b) [Ni(en)2]2+ 與 [Cd(en)2]2+,其中 en = H2NCH2CH2NH2
(c) [Cr(H2O)6]3+ 與 [Mn(H2O)6]2+
6. 在用 FeCl3 與過量的乙二胺四乙酸 (EDTA) 在中性 pH 條件下混合製成的溶液中,Fe3+(aq) 離子的濃度約為 10-17 M。然而,在乙二胺和乙酸濃度相當的溶液中,Fe3+(aq) 濃度約為 10-7,即高出 1010 倍。解釋原因。
7. 配合物 [VO(H2O)5]2+ 為藍色,而與另一種單齒中性配體 L 相似的配合物 [VO(L)5]2+ 為黃色。以下哪些陳述是正確的?簡要解釋。
(a) L 比 H2O 是更強的場配體。
(b) [VO(L)5]2+ 是高自旋配合物。
(c) [VO(L)5]2+ 吸收黃光。
(d) 這兩個配合物都含有一個與金屬相關的 3d 電子。
8. OH- 和 NH3 都是布朗斯臺德鹼,並且都可以與金屬離子形成配合物。解釋為什麼 OH- 可以比 NH3 成為更強的布朗斯臺德鹼,同時在光譜化學序列中卻處於更低的位置。
9. [Ni(H2O)6]2+ 溶液呈淺綠色且順磁性 (µ = 2.90 BM),而 [Ni(CN)4]2- 溶液呈黃色且反磁性。
(a) 繪製每個配合物的分子幾何形狀和 d 軌道能級圖,並顯示 d 軌道的電子佔據情況。
(b) 解釋磁性和顏色的差異。
10. W. Deng 和 K. W. Hipps (J. Phys. Chem. B 2003, 107, 10736-10740) 報道了對 Ni(II) 四苯基卟啉 (NiTPP) 電子性質的 STM 研究,NiTPP 是一種紅紫色、中性的反磁性配合物,透過使 Ni(II) 高氯酸鹽與四苯基卟啉反應制得。當 NiTPP 與硫氰酸鈉反應時,會形成另一種順磁性配合物。繪製 NiTPP 和產物配合物的結構,以及解釋每種結構的晶體場能級圖。你預計順磁性配合物的磁矩值(以 μB 為單位)是多少?

11. 過渡金屬配合物可以發生配體交換反應,其中游離配體或溶劑分子取代了其中一個結合的配體。由於反應物和產物配合物通常具有不同的顏色,因此可以在“試管”反應中輕鬆測量配體交換速率。化學上相同的配體(例如,結合的水分子與遊離的水分子)的交換也可以透過核磁共振波譜和其他方法測量。有趣的是,對於不同的金屬離子和氧化態,水的交換速率變化範圍達 14 個數量級。在某些情況下,一個水分子需要幾周才能與另一個水分子交換。而在其他情況下,交換的時間尺度為納秒。
(a) 存在一個總趨勢(見右側圖),即金屬氧化態越高,交換速率越慢。解釋這種趨勢。晶體場穩定化能與反應動力學有什麼關係?
(b) 除了你的 (a) 中的答案外,解釋你觀察到的二價金屬離子水交換速率的任何趨勢。
(c) Cu2+ 具有特別快的的水交換速率。為什麼?
(d) 圖中交換速率最慢的二價、三價和四價金屬離子的水合配合物的幾何形狀和 d 電子數是多少?它們具有特別高或低的 CFSE 嗎?解釋。
12. 主族離子的配體交換速率沿族向下增加,例如 Al3+ < Ga3+ < In3+。對於過渡金屬離子,我們觀察到相反的趨勢,例如 Fe2+ > Ru2+ > Os2+。解釋為什麼這些趨勢不同。
13. Seppelt 及其同事在鹽 [AuXe4]2+ (Sb2F11-)2 中報道了非常不尋常的離子 [AuXe4]2+(Science 2000, 290, 117-118)。這是首次報道含有金屬與稀有氣體原子之間鍵合的化合物。繪製該離子的 d 軌道能級圖,並預測它應該是反磁性還是順磁性。你預計是否能夠使用 Cu 代替 Au 或 Kr 代替 Xe 來形成類似的配合物?為什麼或為什麼不?
14. 對於反應 順式-Mo(CO)4L2 + CO → Mo(CO)5L + L,發現反應速率對於兩種不同的配體 L 變化了 500 倍,但對 CO 氣體的壓力相對不敏感。(a) 這種反應有什麼型別的機理?(b) 活化體積和活化熵的符號是什麼?
15. 在 Rosenberg 最初發現順式-Pt(NH3)2Cl2 的生物效應時,該化合物是由於 Pt 陽極在含有葡萄糖和氯化鎂的電解質溶液中部分溶解而意外產生的。[27] 電解反應還產生少量銨離子。解釋從機理上講為什麼在這種條件下選擇性地形成順式異構體。
- ↑ Rosi, Nathaniel L.; Eckert, Juergen; Eddaoudi, Mohamed; Vodak, David T.; Kim, Jaheon; O'Keefe, Michael; Yaghi, Omar M. (2003). "微孔金屬有機框架中的儲氫". 科學. 300 (5622): 1127–1129. Bibcode:2003Sci...300.1127R. doi:10.1126/science.1083440. PMID 12750515.
- ↑ Green, M.L.H. (1995). "元素共價化合物的新分類方法". 有機金屬化學雜誌. 500: 127–148. doi:10.1016/0022-328X(95)00508-N.
- ↑ CBC 方法,Parkin 研究組,哥倫比亞大學。
- ↑ Crabtree, Robert. 過渡金屬有機化學:第 4 版。Wiley-Interscience,2005
- ↑ "金屬-配體多重鍵:含氧代、氮化物、亞氨基、亞烷基或烷基炔配體的過渡金屬配合物的化學" W. A. Nugent 和 J. M. Mayer;Wiley-Interscience,紐約,1988。
- ↑ McConville, David H.; Wolf, Jennifer R.; Schrock, Richard R. (1993). "手性鉬 ROMP 引發劑和全順式高度立體規整聚(2,3-(R)2降冰片烯) (R = CF3 或 CO2Me) 的合成". 美國化學會志. 115 (10): 4413–4414. doi:10.1021/ja00063a090.
- ↑ Nguyen, Sonbinh T.; Johnson, Lynda K.; Grubbs, Robert H.; Ziller, Joseph W. (1992). "降冰片烯在質子介質中透過 VIII 族卡賓配合物進行開環複分解聚合 (ROMP)". 美國化學會志. 114 (10): 3974–3975. doi:10.1021/ja00036a053.
- ↑ Rosenberg B,Vancamp L,Trosco JE,Mansour VH(1969)。“鉑化合物——一類新型有效的抗腫瘤藥物”。自然。222(5191):385-386。doi:10.1038/222385a0。
{{cite journal}}:CS1 維護:使用了作者引數(連結) - ↑ G. Wilkinson,M. Rosenblum,M. C. Whiting,R. B. Woodward(1952)。“二茂鐵的結構”。美國化學學會雜誌。74(8):2125-2126。doi:10.1021/ja01128a527。
{{cite journal}}:CS1 維護:作者列表有多個名稱(連結) - ↑ Osborn,J. A.;Jardine,F. H.;Young,J. F.;Wilkinson,G.(1966)。“三(三苯基膦)滷代銠(I)的製備和性質及其一些反應,包括烯烴和炔烴及其衍生物的催化均相氫化”。化學學會雜誌A:1711-1732。doi:10.1039/J19660001711。
{{cite journal}}:CS1 維護:作者列表有多個名稱(連結) - ↑ “三(三苯基膦)滷代銠(I)” J. A. Osborn,G. Wilkinson,《無機合成》,1967年,第10卷,第67頁。DOI 10.1002/9780470132418.ch12
- ↑ D. A. Evans,G. C. Fu和A. H. Hoveyda(1988)。“銠(I)催化的烯烴硼氫化。環狀和非環狀體系中區域和立體化學控制的文獻記錄”。J. Am. Chem. Soc。110(20):6917-6918。doi:10.1021/ja00228a068。
- ↑ I. Ojima,T. Kogure(1972)。“使用矽烷-銠(I)配合物組合選擇性還原α,β-不飽和萜類羰基化合物”。四面體Lett。13(49):5035-5038。doi:10.1016/S0040-4039(01)85162-5。
- ↑ W. S. Knowles(2003)。“不對稱氫化(2001年諾貝爾演講)”。高階合成與催化。345(12):3-13。doi:10.1002/adsc.200390028。
- ↑ J. Yu,J. Long,W. Wu.,P. Xue和X. Zhang(2017)。“銥催化的酮不對稱氫化,使用易得且模組化的二茂鐵基氨基膦酸(f-Ampha)配體”。有機快報。19(3):690-693。doi:10.1021/acs.orglett.6b03862。
{{cite journal}}:CS1 維護:作者列表有多個名稱(連結) - ↑ M. De Santis 等人,造幣金屬(I)氰化物中的化學鍵和s-d雜化,Inorg. Chem. 2019, 58, 11716-11729。https://pubs.acs.org/doi/full/10.1021/acs.inorgchem.9b01694
- ↑ N. Zhang,J. Kou和C. Sun,金浸出配合物中金-配體相互作用的研究:DFT研究,Molecules 2023, 28, 1508。https://doi.org/10.3390/molecules28031508
- ↑ H. Jahn和E. Teller(1937)。“簡併電子態下多原子分子的穩定性。I. 軌道簡併”。皇家學會會刊A。161(905):220-235。Bibcode:1937RSPSA.161..220J。doi:10.1098/rspa.1937.0142。
- ↑ Rob Janes和Elaine A. Moore(2004)。金屬-配體鍵合。英國皇家化學學會。ISBN 0-85404-979-7。
- ↑ Patrick Frank,Maurizio Benfatto,Robert K. Szilagyi,Paola D'Angelo,Stefano Della Longa和Keith O. Hodgson“ [Cu(aq)]2+的溶液結構及其對藍色銅蛋白活性位點中架橋誘導鍵合的影響”無機化學2005,第44卷,第1922-1933頁。DOI 10.1021/ic0400639
- ↑ a b Coe,B. J.;Glenwright,S. J. 八面體過渡金屬配合物中的反式效應。配位化學評論2000,203,5-80。
- ↑ a b Robert H. Crabtree(2005)。過渡金屬有機化學(第4版)。新澤西州:Wiley-Interscience。ISBN 0-471-66256-9。
- ↑ Kauffmann,G. B. 伊利亞·伊里奇·切爾尼亞耶夫(1893-1966)和反式效應。J. Chem. Educ.1977,54,86-89。
- ↑ Chernyaev,I. I. 二價鉑的單亞硝酸鹽。I. Ann. inst. platine(蘇聯)1926,4,243-275。
- ↑ George B. Kauffman,Dwaine O. Cowan(1963)。“順式和反式-二氯二氨合鉑(II)”。《無機合成》(Inorg. Synth)。7:239–245。doi:10.1002/9780470132388.ch63。
{{cite journal}}:CS1 維護:使用了作者引數(連結) - ↑ Helm,Lothar;Merbach,André E.(2005)。“無機和生物無機溶劑交換機制”。《化學評論》(Chem. Rev)。105(6):1923–1959。doi:10.1021/cr030726o。PMID 15941206。
- ↑ Rosenberg,B.;Van Camp,L.;Krigas,T.(1965)。“鉑電極電解產物對大腸桿菌細胞分裂的抑制”。《自然》(Nature)。205(4972):698–9。doi:10.1038/205698a0。PMID 14287410。