
結構生物化學/蛋白質/蛋白質摺疊
蛋白質摺疊是一個多肽摺疊成特定、穩定、功能性、三維結構的過程。它是蛋白質結構獲得其功能性形狀或構象的過程。
蛋白質由長鏈的 氨基酸 構成;它們存在於各種不同的結構中,這些結構通常決定著它們的功能。蛋白質遵循能量有利的途徑來形成穩定的、有序的結構;這被稱為蛋白質的天然結構。大多數蛋白質只有在摺疊後才能執行其各種功能。蛋白質的摺疊途徑或機制是蛋白質為了達到其天然結構而經歷的一系列結構變化。蛋白質摺疊發生在細胞內高度擁擠、複雜、分子環境中,並且為了避免聚集或錯誤摺疊,通常需要分子伴侶的幫助。蛋白質由各種型別的側鏈的氨基酸組成,這些側鏈可能是疏水的、親水的或帶電荷的。這些側鏈的特徵影響了蛋白質將形成的形狀,因為它們在分子內和與周圍環境之間的相互作用方式不同,從而使某些構象和結構比其他構象和結構更有利。科學家認為摺疊蛋白質的指令編碼在序列中。研究人員和科學家可以很容易地確定蛋白質的序列,但還沒有破解控制摺疊的密碼(生命結構 8)。
蛋白質摺疊理論與實驗
[edit | edit source]早期研究 蛋白質組學及其結構的科學家推測,蛋白質具有導致其天然構象的模板。這一理論導致了對蛋白質如何摺疊以獲得其複雜結構的探索。現在眾所周知,在生理條件下,蛋白質通常會自發地摺疊成其天然構象。因此,蛋白質的一級結構是寶貴的,因為它決定了蛋白質的三維結構。通常,大多數生物結構不需要外部模板來幫助它們形成,因此被稱為 自組裝。
蛋白質復性
[edit | edit source]蛋白質復性自 1930 年代就為人所知。然而,直到 1957 年 克里斯蒂安·安芬森 對牛胰 RNase A 進行了一項實驗,蛋白質復性才得以量化。RNase A 是一種單鏈蛋白質,由 124 個殘基組成。在 2-巰基乙醇的 8M 尿素溶液中,RNase A 完全展開,其四個二硫鍵透過還原被裂解。透過對尿素進行 透析並將溶液引入 pH 8 的 O2 中,酶促活性蛋白在物理上無法從 RNase A 中識別出來。因此,該實驗表明蛋白質自發地復性。
RNase A 復性的一個標準是其四個二硫鍵必須重新形成。RNase A 中的八個 Cys 殘基之一與它的天然殘基形成二硫鍵的可能性與其另外七個 Cys 殘基相比是 1/7。此外,剩餘的六個 Cys 殘基中下一個隨機形成下一個二硫鍵的可能性是 1/5,等等。因此,RNase A 隨機重新形成四個天然二硫鍵的機率是 (1/7 * 1/5 * 1/3 * 1/1 = 1/105)。這個機率的結果表明,RNase A 中二硫鍵的形成不是隨機的活動。
當 RNase A 重新氧化利用 8M 尿素時,允許二硫鍵在多肽鏈是無規捲曲時重新形成,那麼當去除尿素後,RNase A 只有大約 1% 的酶促活性。然而,透過使用 2-巰基乙醇,當二硫鍵交換反應發生並且蛋白質回到其天然狀態時,蛋白質可以再次變得完全活躍。RNase A 的天然狀態在生理條件下是熱力學穩定的,特別是由於比天然狀態更穩定的蛋白質需要更大的 活化能壘,並且動力學上無法獲得。
透過使用酶 蛋白質二硫鍵異構酶 (PDI),隨機化的 RNase A 所需的時間縮短到大約 2 分鐘。這種酶有助於促進二硫鍵交換反應。為了使 PDI 能夠發揮作用,它的兩個活性位點 Cys 殘基需要處於 -SH 形式。此外,PDI 幫助蛋白質在獲得熱力學有利構象時隨機裂解和重新形成二硫鍵。
翻譯後修飾的蛋白質可能無法復性
[edit | edit source]處於“混亂”狀態的蛋白質會透過蛋白二硫鍵異構酶 (PDI) 重新摺疊,而處於天然狀態的蛋白質則不需要 PDI,因為天然蛋白質已經處於穩定的構象。然而,經過翻譯後修飾的蛋白質需要二硫鍵來穩定其相對不穩定的天然形式。例如,胰島素 是一種多肽激素。這種由 51 個殘基組成的多肽具有兩個二硫鍵,被 PDI 失活。以下連結顯示了帶有兩個二硫鍵的胰島素影像。透過觀察這種現象,科學家們發現胰島素是由胰島素原 生成的,胰島素原是一種含有 84 個殘基的單鏈。這個連結 提供了更多關於胰島素原結構及其轉化為胰島素過程的資訊。胰島素原的二硫鍵需要在透過蛋白水解切割 其內部的 33 個殘基的 C 鏈之前保持完整。然而,根據兩項發現,C 鏈不是決定 A 和 B 鏈摺疊的關鍵,而是將它們連線在一起,以促進二硫鍵的形成。一方面,在合適的重新摺疊條件下,混亂的胰島素能夠以 30% 的產率恢復到其天然形式。如果 A 和 B 鏈進行交聯,這種產率可以提高。另一方面,透過分析來自許多物種的胰島素原序列,發現 C 鏈的突變率是 A 和 B 鏈的八倍。
蛋白質摺疊的決定因素
[edit | edit source]有各種相互作用有助於穩定天然蛋白質的結構。具體而言,重要的是要研究形成蛋白質結構的相互作用是如何組織的。此外,只有少量的多肽序列可以形成穩定的構象。因此,很明顯,在生物系統中,特定的序列是透過進化而被使用的。
螺旋和片層在蛋白質中占主導地位,因為它們有效地填充空間
[edit | edit source]平均而言,大約 60% 的蛋白質含有大量的α-螺旋 和β-摺疊片。透過疏水相互作用,蛋白質能夠形成緻密的非極性核心,但它們沒有能力指定哪些多肽被限制在特定的構象中。如捲曲形式的多肽片段中所見,氫鍵的數量並不比α-螺旋和β-摺疊片少。這一觀察結果表明,多肽的不同構象形式不受氫鍵需求的限制。 Ken Dill 提出,螺旋和片層的出現是由於凝聚聚合物中空間位阻 造成的。透過實驗和對簡單柔性鏈的構象進行模擬,可以確定β-摺疊片和α-螺旋的比例隨著鏈的複雜程度的增加而增加。因此,可以得出結論,螺旋和片層在蛋白質的複雜結構中很重要,因為它們在蛋白質摺疊中是緻密的。不同力的耦合,例如氫鍵、離子配對和範德華相互作用,進一步促進了α-螺旋和β-摺疊片的形成。
蛋白質摺疊受內部殘基的指導
[edit | edit source]透過研究蛋白質修飾,可以確定不同類別的氨基酸殘基在蛋白質摺疊中的作用。例如,在一項特定研究中,RNase A 的遊離伯氨基被衍生化,與由 8 個殘基組成的多聚-DL-丙氨酸鏈結合。多聚-丙氨酸鏈很大且水溶性,因此可以使 RNase 的 11 個遊離氨基連線起來,而不會干擾蛋白質的天然結構或其重新摺疊的能力。因此,可以得出結論,蛋白質的內部殘基促進了其天然構象,因為 RNase A 的遊離氨基位於外部。此外,研究表明,發生在殘基表面的突變很常見,並且與發生在內部殘基的改變相比,更不可能改變蛋白質的構象。這一發現表明,蛋白質摺疊主要是由疏水力 驅動的。
蛋白質結構是按等級組織的
[edit | edit source]George Rose 證明了蛋白質結構域由亞結構域組成,並且進一步具有亞亞結構域等等。因此,很明顯,大型蛋白質具有連續的、緻密的、物理上可分離的結構域。當將天然蛋白質中的多肽片段視覺化為一條帶有許多纏結的繩子時,在將繩子切成兩段時可以看到一個平面。當用藍色和紅色突出顯示 n 個殘基結構域中的 n/2 個殘基時,可以重複此過程。隨著此過程的重複,可以看出,在所有階段,蛋白質的紅色和藍色區域都不會相互滲透。以下連結 顯示了 HiPIP(高電位鐵蛋白)的 X 射線結構及其前 n/2 個殘基在 n 個殘基蛋白質上的顏色分別為紅色和藍色。此外,第二行和第三行中顯示的後續結構顯示瞭如所示的 n/2 殘基分裂的重複過程,其中蛋白質的左側具有其第一個和最後一個一半,分別為紅色和藍色,而鏈的其餘部分為灰色。透過這個例子,可以清楚地看到蛋白質結構是按等級組織的,這意味著多肽鏈被視為亞結構域,它們本身是緻密的結構,並與相鄰的結構相互作用。這些相互作用形成了一個更大的井然有序的結構,這主要是由於氫鍵相互作用,並且在理解多肽 如何摺疊形成其天然結構方面起著重要的作用。
蛋白質結構是可適應的
[edit | edit source]由於側鏈 在球狀蛋白質 內部以高度互補的方式相互配合,因此其堆積密度幾乎與有機晶體相同。因此,為了確認這種高堆積密度現象是否是影響蛋白質結構的重要因素,Eaton Lattman 與 George Rose 試圖驗證球狀蛋白質中是否存在側鏈之間的相互作用。他們分析了 67 種經過充分研究的球狀蛋白質結構,並得出結論,沒有發現偏好的相互作用。該實驗表明,堆積不是決定天然摺疊的原因,而是天然摺疊是球狀蛋白質堆積的必要條件。這一觀點可以得到進一步的支援,因為蛋白質家族的成員即使沒有序列相似性和遠緣關係,也會產生相同的摺疊。
此外,結構實驗資料表明,蛋白質內部殘基以有效的方式緊密結合在一起有各種方式。布萊恩·馬修斯 基於T4溶菌酶(由 噬菌體T4 產生)進行了一項廣泛的研究,結果發現T4溶菌酶殘基的變化只會影響區域性位移,不會導致任何全域性結構變化。以下 連結 提供了T4溶菌酶的X射線檢視和對其結構的簡要生化描述。馬修斯研究了164個殘基的T4溶菌酶的超過300種不同突變體,並將它們進行比較。此外,還觀察到T4溶菌酶可以承受大約4個殘基的插入,同時仍然不會對整體蛋白質結構或酶活性產生任何重大結構變化。此外,透過使用測定技術表明,在進行的2015個單殘基替換中,只有173個T4突變體表現出酶活性顯著降低。透過這些實驗,可以明顯看出蛋白質結構具有極強的耐受性。
萊文薩爾悖論
[edit | edit source]萊文薩爾悖論是一個思想實驗,也是蛋白質摺疊理論中的自指。1969年,賽勒斯·萊文薩爾注意到,由於展開的多肽鏈具有非常多的自由度,因此該分子具有天文數量的可能構象。在他的一篇論文中估計了3300 或10143 個構象。
萊文薩爾悖論觀察到,如果蛋白質透過依次取樣所有可能的構象來摺疊,那麼即使以快速速率取樣構象,也需要花費大量時間才能完成摺疊。基於蛋白質摺疊速度遠快於此的觀察結果,萊文薩爾隨後提出,不會發生隨機構象搜尋,因此蛋白質必須透過一系列亞穩態中間狀態來摺疊。
1969年,賽勒斯·萊文薩爾計算出,如果蛋白質要從展開狀態到天然狀態摺疊時隨機取樣所有可能的構象,即使蛋白質每秒達到1000億個構象,也將需要天文時間。觀察到蛋白質在相對較短的時間內摺疊,萊文薩爾提出蛋白質以固定且定向的過程摺疊。我們現在知道,雖然蛋白質摺疊不是一個隨機過程,但似乎並沒有單一的固定蛋白質摺疊途徑。這一觀察被稱為萊文薩爾悖論。這個悖論清楚地表明,蛋白質不會透過嘗試所有可能的構象來摺疊。相反,它們必須遵循至少部分定義的摺疊路徑,該路徑由完全變性蛋白與其天然結構之間的中間體組成。
累積選擇
[edit | edit source]擺脫萊文薩爾悖論的關鍵在於認識到累積選擇。理查德·道金斯曾問,一隻猴子隨機敲擊打字機需要多長時間才能打出“Methinks it is like a weasel”,這是哈姆雷特對波洛尼烏斯的一句臺詞。需要大量擊鍵,約為1040 個。然而,如果我們假設每個正確的字元都保留下來,只讓猴子重新打出錯誤的字元,平均只需要幾千次擊鍵。這兩種情況之間的關鍵區別在於,第一種情況使用完全隨機搜尋,而第二種情況保留了部分正確的中間體。這也表明蛋白質摺疊的本質是保留部分正確的中間體,儘管蛋白質摺疊問題比莎士比亞例子中提出的問題要困難得多。
成核-凝聚模型
[edit | edit source]為了正確理解蛋白質摺疊問題,我們必須考慮蛋白質的某些特徵。由於蛋白質只有輕微的穩定性,因此典型1000個殘基的蛋白質的摺疊狀態和展開狀態之間的自由能差為42 kJ mol−1,因此每個殘基平均只貢獻0.42 kJ mol−1 的能量來維持摺疊狀態。這個能量值小於室溫下的熱能,即2.5 kJ mol−1。這種微不足道的穩定能量意味著正確的中間體,尤其是摺疊早期形成的中間體,可能會丟失。然而,導致協同摺疊的相互作用可以隨著結構的建立而穩定中間體。因此,具有顯著結構偏好的區域性區域,雖然它們本身可能不穩定,但往往會採用其偏好的結構,並且隨著它們的形成,可以相互作用,導致穩定性的提高。成核-凝聚模型指的是解決蛋白質摺疊挑戰的這個概念框架。
分子內相互作用在摺疊機制中的作用
[edit | edit source]蛋白質 摺疊形成能量上有利的結構,這些結構由 疏水相互作用 聚集、 氫鍵 和 範德華力 在 氨基酸 之間的相互作用所穩定。蛋白質摺疊首先形成二級結構,如α螺旋、β摺疊和環。不同的氨基酸具有不同的傾向,它們會根據氨基酸的極性和旋轉勢壘而形成α螺旋、β摺疊或β轉角。例如,纈氨酸、蘇氨酸、異亮氨酸等氨基酸由於空間位阻會導致α螺旋不穩定。因此,它們更傾向於向β摺疊而不是α螺旋發生構象變化。氨基酸在二級結構中的相對頻率根據它們對α螺旋、β摺疊或轉角的偏好進行分組(表1)。 表1:氨基酸殘基在二級結構中的相對頻率 這些結構反過來又摺疊形成三級結構,並透過形成分子內氫鍵來穩定。 共價 鍵也可能在摺疊成三級結構期間透過形成二硫鍵或金屬簇而發生。根據羅伯特·佩恩的“蛋白質摺疊機制”,分子也經常會透過形成疏水坍塌(其中所有疏水側鏈突然滑入蛋白質內部或聚集在一起)形成的中間“熔融球狀體”狀態,然後達到其天然構象。然而,這意味著所有主鏈NH和CO基團都被埋藏在非極性環境中,但它們更喜歡水性環境,因此二級結構必須很好地配合在一起,以便透過氫鍵和 範德華力 相互作用的穩定性超過它們的親水性傾向。氫鍵在蛋白質中的強度會根據它們在結構中的位置而變化;在疏水核心形成的氫鍵比暴露於水性環境中的氫鍵對天然狀態的穩定性貢獻更大。
水溶性蛋白質摺疊成具有非極性疏水核心的緊湊結構。蛋白質內部包含中心非極性殘基(例如-亮氨酸、纈氨酸、甲硫氨酸和苯丙氨酸),而外部主要包含極性帶電殘基(例如-天冬氨酸、穀氨酸、賴氨酸和精氨酸)。這樣,極性帶電分子就可以與周圍的水分子相互作用,而疏水分子可以免受水性環境的包圍。最小化結構外部的疏水側鏈數量使蛋白質結構在熱力學上更有利,因為疏水分子更喜歡在水性環境中聚集在一起(例如-疏水效應)。跨越生物膜的蛋白質(例如-孔蛋白)具有與水溶性天然結構相反的內外分佈,它們具有疏水殘基覆蓋的外部表面,中心充滿水,襯裡是帶電和極性氨基酸。
膜蛋白的摺疊
[edit | edit source]在“摺疊場景調查:膜蛋白”這篇文章中,作者帕梅拉·J·布斯和保羅·庫爾諾試圖回答具有α螺旋結構的跨膜蛋白的摺疊機制是如何運作的。研究膜蛋白的摺疊一直很困難,因為這些蛋白通常很大,並且由多個亞基組成。這些蛋白具有高度的構象靈活性,這對它們在細胞中發揮作用是必要的。此外,這些蛋白既有疏水錶面,面向膜,也有親水錶面,面向膜兩側的水性區域。這些蛋白在側向移動,並共享其嵌入的脂質雙層的彈性特性。為了研究這些蛋白,布斯和庫爾諾認為必須操縱脂質雙層,並結合動力學和熱力學研究方法。
可逆折疊和線性自由能 蛋白質摺疊的自由能透過可逆化學變性來衡量。蛋白質的可逆折疊取決於這種自由能。對於正在研究的α螺旋蛋白質,已經證明它們遵循可逆的二態過程。bR(一種稱為細菌視紫紅質的α螺旋膜蛋白)在將SDS(一種變性劑,也是一種陰離子去垢劑)新增到混合脂質、去垢劑膠束中時會可逆地展開。該二態反應涉及部分展開的SDS狀態和摺疊的bR狀態。透過比較展開和摺疊速率的對數以及SDS摩爾分數,產生了線性圖,證明了線性關係。該圖證明bR在膜外具有非常高的穩定性,證明它出乎意料地穩定。此外,bR在膜外非常穩定,在沒有新增變性劑的情況下,它不會在合理的時間段內展開。
與水溶性蛋白質的比較 Booth和Curnow研究了三種資訊量最多的膜蛋白:bR、DGK(大腸桿菌二醯基甘油激酶)和KcsA(鏈黴菌利迪亞斯鉀通道)。這三種膜蛋白與水溶性蛋白質(透過2或3態動力學摺疊)進行了比較。在沒有變性劑的情況下,展開的總自由能變化對於水溶性蛋白質和類似大小的膜蛋白是相同的。這證明決定蛋白質穩定性的不是力的型別,而是弱力的平衡。事實證明,膜蛋白中的氫鍵與水溶性蛋白質中的氫鍵強度相似,而不是像預期的那樣在膜蛋白中更強。
機械強度和在施加力下的展開 動態力顯微鏡可用於測量蛋白質特定區域在施加力下的機械響應。在這種情況下,展開力取決於活化能壘。這種展開與蛋白質的熱力學穩定性無關。對於在施加力下的展開,膜蛋白(尤其是bR)似乎遵循哈蒙德行為規則。該反應兩個連續狀態之間的能量差減小,狀態在結構上變得相似。
周圍膜的影響 膜蛋白受其周圍膜的影響很大。如果脂質在去垢劑膠束中摻入,從而增加脂質結構的穩定性,則蛋白質及其摺疊都將被穩定。不同脂質的不同組合會導致膜蛋白的不同穩定性或摺疊。膜的大小也會影響膜蛋白。不同型別的脂質會導致不同的膜特性。一種稱為PE脂質的脂質比第二種稱為PC脂質的脂質具有更高的自發曲率。透過將PE脂質新增到PC脂質中,雙層的單層曲率增加。增加脂質雙層的曲率會增加蛋白質摺疊的穩定性。
蛋白質在生物膜中的轉運
[edit | edit source]在線粒體中,由核糖體制造的蛋白質直接從胞質溶膠中吸收。線粒體蛋白首先在胞質溶膠中完全合成,作為線粒體前體蛋白,然後被攝取到膜中。線粒體蛋白在其N端含有特定的訊號序列。這些訊號序列通常在進入膜後被去除,但進入具有外膜、內膜、膜間空間的膜的蛋白質具有內部序列,這些序列在內膜內的轉運中起主要作用。
蛋白質轉運在將蛋白質轉運穿過線粒體膜中起主要作用。在外膜和內膜中發現了四個主要的多分支蛋白複合物。TOM複合物位於外膜,兩種型別的TIM複合物整合在內膜中:TIM23和TIM22。這些複合物充當線粒體前體蛋白的受體。
TOM:匯入所有細胞核編碼的蛋白質。它主要啟動訊號序列向膜間空間的轉運,並將跨膜蛋白插入外膜空間。然後,一個稱為SAM複合體的β桶複合物負責在外膜中正確摺疊蛋白質。TIM23位於內膜,調節可溶性蛋白質插入基質,並促進跨膜蛋白質插入內膜。TIM23是另一個內膜複合物,促進由轉運蛋白組成的內膜蛋白的插入,這些轉運蛋白線上粒體膜之間轉運ADP、ATP和磷酸鹽。OXA是另一個內膜複合物,幫助插入線粒體本身合成的內膜蛋白,以及插入首先被轉運到基質空間的內膜蛋白。File:Translocation.jpg
在核糖體上摺疊
[edit | edit source]蛋白質鏈開始摺疊的地方是一個被廣泛研究的主題。當新生鏈穿過核糖體的“出口隧道”進入細胞環境時,鏈何時開始摺疊?將討論核糖體隧道中協同翻譯摺疊的概念。蛋白質的新生鏈透過其C端與肽基轉移酶中心(PTC)結合,並將以向量方式出現。隧道非常狹窄,對新生鏈施加了一定的剛性,隨著每個氨基酸的新增,蛋白質的構象空間增加。協同翻譯摺疊可以幫助透過幫助蛋白質在仍然位於核糖體隧道中時獲得顯著水平的天然狀態來大大減少可能的構象空間。蛋白質的長度也可以很好地估計其三維結構。較小的鏈傾向於有利於β摺疊,而較長的鏈(如達到119/153個殘基的鏈)傾向於有利於α螺旋。
核糖體隧道長度超過80Å,寬度約為10-20Å。在隧道內部,有輔助分子,如L23、L22和L4蛋白質,它們與新生鏈相互作用,幫助摺疊。隧道也具有親水性,並幫助新生鏈穿過它,不會受到阻礙。儘管很硬,但隧道不是被動的導管,但它是否具有促進蛋白質摺疊的能力尚不清楚。最近一項涉及冷凍電鏡的實驗表明,隧道中存在摺疊區。在出口口(距PTC約80Å),新生鏈已採用優選的低階構象。這強化了鏈可以在某些區域具有摺疊程度的建議。儘管可以發生一些低階摺疊,但天然狀態的採用發生在隧道外,但並不一定是在新生鏈被釋放時。結合的新生鏈(RNC)採用部分摺疊結構,在擁擠的細胞環境中,這會導致鏈自締合。然而,這種自締合通過沿著出口隧道排列的交錯核糖體得到緩解,這最大限度地增加了RNC之間的距離。
生成用於研究的RNC
一種生成RNC並拍攝其從隧道中出現的快照的技術是停止翻譯。使用沒有終止序列的截短的DNA。這允許新生鏈保持繫結,直到需要為止。為了確定鏈的殘基,可以透過碳-13或氮-15對其進行標記,然後透過核磁共振波譜檢測。另一種技術是PURE方法,它包含翻譯所需的最小成分。這種方法已被用於研究鏈與輔助分子(如TF伴侶)之間的相互作用。該方法與石英晶體微天平技術相結合,用於透過質量分析合成。生成RNC鏈的體內技術可以透過在高細胞密度下刺激它來完成。這最初是在未標記的環境中完成的,然後將細胞轉移到標記的培養基中。RNC由SecM生成。透過親和層析純化RNC,並透過SDS-PAGE或免疫印跡檢測。
透過生成RNC,可以進行許多實驗來更多地研究出現的新生鏈。如上所述,鏈以向量方式從出口隧道中出現。這使鏈能夠對天然摺疊進行取樣,並增加了摺疊到天然狀態的機率。除了這種向量摺疊之外,伴侶還有助於有利的摺疊速率和正確的摺疊。
內質網中的蛋白質摺疊
[edit | edit source]蛋白質進入哺乳動物內質網:內質網(ER)是蛋白質成熟的主要檢查點,以確保只有正確摺疊的蛋白質被分泌並遞送到作用部位。蛋白質進入內質網始於識別N'端訊號序列。特別地,這種序列被訊號識別蛋白(SRP)檢測到,導致核糖體/新生鏈/SRP複合物與內質網膜結合。然後,該複合物透過一個稱為Sec61轉運蛋白的蛋白質孔,該孔允許多肽鏈進入內質網的腔室部分。
蛋白質摺疊過程中相互衝突的過程:蛋白質進入內質網後,蛋白質分解成摺疊中間體的集合。這些中間體採取三種不同的路線。它們要麼被正確摺疊並被髮送到從內質網(ER)輸出到胞質溶膠中,要麼被聚集起來,要麼被挑選出來降解。這三個過程在競爭中正確分泌蛋白質。為了使蛋白質被正確分泌,摺疊、聚集和降解之間的競爭必須有利於摺疊,以便摺疊比其他過程更快地發生。這種平衡被稱為蛋白穩態。可以透過使用更小的分子來穩定蛋白質(稱為輔因子)或增加摺疊因子的濃度來將蛋白穩態的平衡傾斜為有利於摺疊。這種控制蛋白穩態的能力使科學家能夠克服一些蛋白質摺疊疾病,如囊性纖維化。
正確摺疊的蛋白質已準備好進行順行轉運,並透過內質網膜分泌到胞質溶膠中,方法是透過識別正確摺疊的蛋白質的貨物受體。摺疊不正確的蛋白質不會被分泌,要麼被靶向降解,要麼被聚集。聚集的蛋白質能夠重新進入蛋白質集合的階段,準備摺疊,以便它們可以再次嘗試正確摺疊。

內質網中的摺疊因子
關於摺疊途徑的生化研究提供了一個關於內質網中蛋白質摺疊所涉及的摺疊因子或伴侶的綜合清單。摺疊因子根據它們是否催化某些步驟或它們是否與摺疊途徑中的中間體相互作用進行分類。通常將一般的蛋白質摺疊因子分為四組:熱休克蛋白作為伴侶或伴侶蛋白、肽醯脯氨醯順式/反式異構酶(PPIases)、氧化還原酶和糖基結合蛋白。
許多摺疊因子之所以很棒是因為它們具有多功能性。一個摺疊因子可以處理摺疊途徑的不同區域。不幸的是,這會導致冗餘,因為不同型別的蛋白質執行重疊的功能。這種功能冗餘使得難以理解單個摺疊因子在幫助客戶蛋白成熟中的具體作用。摺疊因子也傾向於在成熟過程中協同作用,這進一步模糊了每個因子的個體作用。由於這些作用尚不清楚,因此很難確認即使一個摺疊因子處理了一個蛋白質中的特定反應,該摺疊因子也會在另一個蛋白質中執行相同的函式。
除了幫助蛋白質的非共價摺疊和展開外,內質網中的摺疊因子有時會延遲與蛋白質的相互作用。這為新生蛋白質正確摺疊提供了時間,並使摺疊的蛋白質能夠在其摺疊途徑上回溯,這延長了未摺疊狀態的平衡,防止蛋白質被保持在非天然狀態。
內質網後的摺疊:雖然內質網只提供正確組裝的蛋白質以分泌,但也有一些例子表明蛋白質在高爾基體和更遠的地方改變構象。通常,新摺疊的蛋白質在內質網中很敏感且易於展開,但在退出後抗展開。在沒有伴侶蛋白和其他摺疊酶的環境中,蛋白質是緊湊的,並且在退出內質網後對改變相對抵抗。然而,這並不一定意味著蛋白質摺疊結束,因為一些分子伴侶蛋白如Hsp 70s和Hsp 90s在蛋白質存在的整個過程中繼續幫助蛋白質構象。

研究蛋白質摺疊的一種策略是在高濃度的化學變性劑(如鹽酸胍)中展開蛋白質分子。然後迅速稀釋溶液,直到變性劑濃度降低到使天然狀態再次熱力學穩定。之後,可以觀察蛋白質摺疊的結構變化。理論上,這聽起來很簡單。然而,此類實驗很複雜,因為展開的蛋白質在化學變性劑中具有無規捲曲狀態。此外,分析樣品中發生的結構變化可能很困難,因為所有分子可能具有顯著不同的構象,直到反應的最後階段。因此,分析必須在幾秒鐘內完成,而不是通常允許推斷單個天然蛋白質構象結構的天數或數週。為了避免這個問題,可以在蛋白質展開後還原二硫鍵,並在氧化條件下重新形成。然後,可以透過質譜等標準技術識別蛋白質,以得出有關二硫鍵形成時的摺疊階段存在的結構的結論。
多種技術用於監測重摺疊過程中的結構變化。例如,在圓二色性中,遠距離的紫外光用於提供摺疊過程中二級結構出現的測量。近距離的紫外光監測芳香殘基緊密堆積環境的形成。 核磁共振也是一種有用的技術,可以表徵單個氨基酸殘基水平的構象。它還可以用於監測結構的形成如何保護醯胺氫免受溶劑交換的影響。
圓二色性:這種型別的光譜法測量圓偏振光的吸收,因為蛋白質的結構,例如α螺旋和β摺疊,是手性的,可以吸收這種光。光的吸收指示蛋白質摺疊程度。這種技術還可以透過測量吸光度隨變性劑濃度或溫度的變化來測量蛋白質的平衡展開。變性劑熔解測量展開的自由能,而溫度熔解測量蛋白質的熔點。這種技術是研究蛋白質摺疊最普遍和最基本的方法。
雙偏振干涉測量法:這種技術使用限制在波導中的雷射束的倏逝波來探測已吸收到波導表面的蛋白質層。雷射光聚焦在兩個波導上,一個感知光束並具有暴露的表面,另一個用於建立參考光束並激發波導的偏振模式。對干涉圖的測量可以幫助計算蛋白質密度或摺疊,吸收層的尺寸,並推斷關於亞原子解析度的分子相互作用的結構資訊。當透過兩個波導的光結合時,在遠場中獲得二維模式。
質譜法:使用 質譜法 研究蛋白質摺疊的優勢包括能夠檢測具有不同氘含量的分子,這使得能夠研究蛋白質摺疊反應的異質性。它還可以測量與分子伴侶蛋白結合的摺疊中間體的構象,而不會破壞複合物。質譜法還可以直接比較重摺疊特性,因為如果兩種蛋白質的分子量足夠不同,則可以在沒有分離的情況下研究蛋白質混合物。
高時間解析度:這些是快速時間分辨技術,其中展開的蛋白質樣品被觸發快速摺疊。然後研究產生的動力學。實現這一點的方法包括快速混合溶液、光化學方法和雷射溫度跳躍光譜法。
蛋白質三級結構的計算預測:這是一種與蛋白質摺疊相關的蛋白質結構分析的不同形式。這些程式可以模擬冗長的摺疊過程,提供統計勢的資訊,並再現摺疊途徑。
蛋白質錯誤摺疊是指蛋白質未能有效地實現其緊密堆積的天然構象,或者由於環境變化或突變導致穩定性降低而未能維持該構象。已經確定,蛋白質摺疊失敗是在高溫和其他壓力情況下的一種普遍現象。錯誤摺疊蛋白質的兩個最常見結果是降解和聚集。當多肽從細胞中出現時,它可能會摺疊成天然狀態、被蛋白水解降解或與其他分子形成聚集體。蛋白質處於不斷的動態平衡狀態,因此即使摺疊過程已完成,在細胞環境中也會發生展開。展開的蛋白質通常會重新摺疊回其天然狀態,但如果控制過程失敗,錯誤摺疊會導致細胞功能障礙,並最終導致疾病。與錯誤摺疊相關的疾病涵蓋多種病理狀況,例如囊性纖維化,其中編碼結果的基因發生突變,導致摺疊成一種構象,其分泌被細胞中的質量控制機制阻止。大約 50% 的癌症與 p53 蛋白的突變有關,最終導致細胞週期控制的喪失,並導致腫瘤生長。蛋白質無法保持摺疊會導致聚集,這是被稱為澱粉樣蛋白沉積症的一組遺傳性、散發性和感染性疾病的常見特徵。聚集通常導致無序的物質,可以在生物體中降解,但它也可能導致在組織中積累的高度不溶性纖維。已知大約有 20 種疾病是由澱粉樣蛋白物質的形成引起的,包括阿爾茨海默病、II 型糖尿病和帕金森病。澱粉樣蛋白纖維是有序的蛋白質聚集體,由於分子間氫鍵具有廣泛的β摺疊結構,並且與它們衍生的蛋白質具有總體相似的外觀。澱粉樣蛋白纖維的形成是由於長時間暴露於至少部分變性的條件下。
阿爾茨海默病:這種神經退行性疾病是由 斑塊 和 纏結 在大腦神經細胞中積累引起的。[1] 斑塊幾乎完全由一種蛋白質組成,是β-澱粉樣蛋白在神經細胞間隙之間聚集,而纏結是tau蛋白在神經細胞內部聚集。纏結在廣泛的神經細胞疾病中很常見,而神經炎斑塊更特異於阿爾茨海默病。儘管科學家不確定斑塊和纏結在阿爾茨海默病形成中起什麼作用,但一種理論是,這些積累的蛋白質阻礙了神經細胞彼此通訊的能力,並使其難以存活。研究表明,斑塊和纏結在人們衰老時自然發生,但在患有阿爾茨海默病的人群中觀察到更多。這種增加的原因尚不清楚。
克雅氏病(瘋牛病):這種疾病是由稱為朊病毒的異常蛋白質引起的,它們會侵蝕大腦並形成洞狀病變。朊病毒(proteinaceous infectious virion)被發現是具有改變構象的蛋白質。科學家推測,這些感染性因子可以與其他類似蛋白質結合,並誘導它們的構象發生改變,從而傳播新的感染性蛋白質。[2] 朊病毒對熱、紫外線和輻射高度耐受,這使得它們難以消除。在克雅氏病中,潛伏期長達數年,然後迅速發展為抑鬱、行走困難、痴呆和死亡。目前還沒有針對朊病毒病的有效治療方法,所有朊病毒病都是致命的。[3]
帕金森病:編碼α-突觸核蛋白的基因發生突變是某些罕見家族性帕金森病的病因。迄今已發現三種點突變:A53T、A30P 和 E46K。此外,該基因的重複和三聯體重複也可能是其他帕金森病血統的病因。帕金森病患者的主要症狀是由於基底神經節對運動皮層的刺激減少,而基底神經節的刺激通常是由多巴胺的不足產生和作用引起的。多巴胺是在大腦的多巴胺能神經元中產生的。患有這種疾病的人會發生腦細胞丟失(多巴胺能神經元死亡),這可能是由異常積累的蛋白質α-突觸核蛋白與受損細胞中的泛素結合造成的。這使得α-突觸核蛋白-泛素複合物無法被引導到蛋白酶體。新的研究表明,內質網和高爾基體之間蛋白質的錯誤運輸可能是α-突觸核蛋白導致多巴胺能神經元丟失的原因。
囊性纖維化:弗朗西斯·柯林斯於 1989 年首次發現了這種遺傳性基因突變。問題出現在囊性纖維化跨膜傳導調節因子 (CFTR)中,該調節因子調節鹽含量並防止細菌生長,當 CFTR 的解離被擾亂時,CFTR 作為一種蛋白質調節跨細胞膜的氯離子轉運。[4] 缺失的氨基酸無法殺死肺部的細菌,從而導致慢性肺部感染,最終導致早逝。[5] 科學家已經使用核磁共振波譜 (NMR)來研究囊性纖維化及其影響。
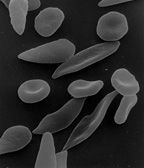
鐮狀細胞貧血:鐮刀狀紅血球粘附在狹窄的血管壁上,阻礙血液流動,這就是鐮狀細胞貧血的定義。血液中紅血球數量不足以及攜氧血液缺乏會導致嚴重的醫療問題。透過檢測兩個缺陷的遺傳基因的存在,可以檢測到血紅蛋白基因的缺陷。當血紅蛋白釋放氧氣時,就會形成鐮刀狀形狀,從而導致僵硬的紅血球形成桿狀結構。一些症狀包括:疲勞、呼吸急促、任何關節或身體器官的疼痛持續時間不等、眼部問題可能導致失明、皮膚和眼睛發黃,這是由於紅血球快速分解造成的。幸運的是,鐮狀細胞貧血可以透過簡單的血紅蛋白電泳血液測試檢測出來。儘管目前還沒有治癒方法,但輸血、口服抗生素和羥基脲可以減輕疼痛。[6]
亨廷頓病:也稱為三核苷酸重複病,亨廷頓病是由亨廷頓蛋白中谷氨醯胺重複造成的。大約 40 個或更多個 C-A-G(谷氨醯胺)的重複會導致亨廷頓病,因為正常數量在 10 到 35 個之間。在突變的亨廷頓蛋白 (mHTT) 的翻譯後修飾過程中,一小部分多聚谷氨醯胺擴充套件會錯誤摺疊形成包涵體。包涵體對腦細胞有毒。這種亨廷頓蛋白的改變沒有明確的影響,只是它影響神經細胞的功能。[7] 這種無法治癒的疾病影響肌肉協調和一些認知功能。
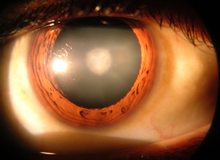
白內障:眼晶狀體由稱為晶狀體蛋白的蛋白質組成。晶狀體蛋白在晶狀體細胞質中具有果凍狀的質地。白內障是目前世界上導致失明的主要原因,當晶狀體蛋白分子形成聚集體,散射可見光,導致眼晶狀體變得渾濁時就會發生白內障。紫外線和氧化劑被認為會導致白內障,因為它們可能會改變晶狀體蛋白的化學結構。在兒童中,人們觀察到 αB-晶狀體蛋白的缺失或突變會促進白內障的形成。隨著年齡的增長,患白內障的可能性呈指數級增長。Pain, Roger H. (2000). 蛋白質摺疊機制. 牛津大學出版社. pp. 420–421. ISBN 019963788. Retrieved 2009-10-18. {{cite book}}: Check |isbn= value: length (help)
澱粉樣蛋白纖維。
[edit | edit source]蛋白質錯誤摺疊是由摺疊效率下降引起的,這會導致可用於執行其正常作用的蛋白質數量減少,並形成澱粉樣蛋白纖維,即聚集的蛋白質結構,形成交叉β結構,可以產生許多生物學功能。蛋白質聚集可以來自翻譯後發生的各種過程,包括內質網 (ER) 質量控制系統中降解可能性增加、蛋白質運輸不當,或特定肽和蛋白質從其可溶性功能狀態轉換為其高度組織化的聚集纖維。
結構。
X 射線晶體學。
透過 X 射線晶體學,形成了澱粉樣蛋白纖維結構的三維晶體,並檢查了肽的形成結構以及分子如何堆積在一起。在一個特定的片段中,發現晶體包含平行 β-摺疊片的部分,其中每個肽貢獻一個單一的 β-鏈。β-鏈堆疊在一起,形成的 β-摺疊片是平行的,側鏈 Asn2、Gln4 和 Asn6 以一種使水從兩個 β-摺疊片之間的區域排除的方式相互作用,而其餘的側鏈在外部被水合並遠離下一個 β-摺疊片。
固態核磁共振 (SSNMR)。
透過固態核磁共振 (SSNMR) 以及其他方法的幫助,例如計算能量最小化、電子順磁共振以及定點熒游標記和氫氘交換、質譜、有限蛋白水解和脯氨酸掃描誘變,澱粉樣蛋白纖維的結構被認為是四個β-摺疊片,相隔約 10Å。
透過 NMR 與計算能量最小化,確定在 pH 7.4 和 24 ˚C 下澱粉樣β肽的 40 個殘基形式為纖維的核心貢獻了一對 β-鏈,該 β-鏈透過一個蛋白質環連線。澱粉樣β肽以平行方式相互堆疊。
從定點自旋標記與電子順磁共振 (SDSL-EPR) 實驗中發現,該分子在纖維中結構非常完整,並且排列平行。SDSL-EPR 以及氫氘交換、質譜、有限蛋白水解和脯氨酸掃描誘變表明,該結構在 N 末端側具有高靈活性並暴露於溶劑,但在結構的其他部分則很剛性。
透過 SSNMR 與熒游標記和氫氘交換進行的實驗確定,C 末端參與了纖維結構的核心,每個分子貢獻了四個 β-鏈,其中鏈 1 和 3 形成一個 β-摺疊片,鏈 2 和 4 形成另一個 β-摺疊片,相隔約 10Å。

使用 SSNMR 技術進行的進一步實驗在原子水平上進行了研究,結果在光譜中產生了非常窄的共振線,表明纖維中的分子具有某種程度的均勻性,肽顯示出擴充套件的 β-鏈與纖維相連。
結論。
透過 X 射線晶體學或 SSNMR 確定的結構與先前從胰島素形成的冷凍電子顯微鏡 (EM) 中提出的結構類似。EM 利用電子密度圖,揭示了結構中未扭曲的 β 摺疊。這些實驗中發現的結構相似性表明許多澱粉樣纖維可能具有相似的特徵,例如側鏈堆積、β 鏈對齊和 β 摺疊的分離。 [8] Annu. Rev. Biochem. 2006.75:333-366. www.annualreviews.org. Retrieved 24 Oct 2011</ref>
形成
形成被認為具有遺傳性的澱粉樣蛋白結構的能力源於發現越來越多蛋白質沒有顯示與蛋白質相關的疾病跡象。人們發現,澱粉樣蛋白可以從具有功能而不是疾病相關特徵的自身蛋白質轉化而來,存在於活生物體中。
在這些蛋白質突變中,影響澱粉樣纖維形成的不同因素和不同的鏈以不同的速度形成澱粉樣纖維。在不同的多肽分子中,疏水性、親水性、電荷變化、溶劑暴露程度、芳香族側鏈數量、表面積和偶極矩都會影響蛋白質的聚集速度。人們發現,蛋白質濃度、溶液的 pH 值和離子強度以及蛋白質所在的氨基酸序列決定了從非結構化、非同源蛋白質序列中聚集的速度。
側鏈疏水性的增加或減少會改變蛋白質聚集的傾向。
蛋白質中的電荷可以透過多肽鏈與周圍其他大分子相互作用而產生聚集。此外,β 摺疊形成的低傾向性以及 α 螺旋形成的高傾向性也有助於澱粉樣蛋白的形成。
人們發現,蛋白質序列暴露於溶劑的程度往往會影響澱粉樣蛋白的形成。暴露於溶劑的蛋白質似乎會促進聚集。即使蛋白質的其他部分具有較高的聚集傾向,但並沒有參與聚集,它們似乎至少部分未暴露於溶劑,而暴露於溶劑但未參與聚集的其他區域則具有較低的形成澱粉樣纖維的傾向。
甚至有人提出,蛋白質序列隨著時間的推移而進化,透過交替疏水和親水區域的模式來避免形成疏水殘基簇,從而降低蛋白質聚集發生的傾向。 [8]
序列對澱粉樣蛋白形成的影響
澱粉樣蛋白的形成主要來自所有肽和蛋白質中相似的多肽鏈的特性,但有時序列會影響分子構象狀態的相對穩定性。在這種情況下,具有不同序列的多肽鏈會以不同的速度形成澱粉樣纖維。序列差異會影響蛋白質聚集的行為,而不是影響蛋白質摺疊的穩定性。各種物理化學因素會影響未摺疊多肽鏈澱粉樣結構的形成。
側鏈的疏水性會影響未摺疊多肽鏈的聚集。當聚集位點區域的氨基酸在突變或摺疊位點增加或減少疏水性時,它們會改變序列的聚集能力。隨著時間的推移,序列已經進化來避免產生疏水殘基團塊,透過交替蛋白質的疏水區域。
電荷會影響澱粉樣蛋白摺疊的聚集。高淨電荷可能具有阻礙蛋白質自締合的可能性。減少正淨電荷的突變可能導致與增加正淨電荷相反的聚集形成效果。人們發現,多肽鏈可以透過與高電荷大分子相互作用來執行,這表明蛋白質聚集的電荷很重要。
蛋白質的二級結構也會影響澱粉樣蛋白的聚集。研究表明,形成 α 螺旋結構的低機率和形成 β 摺疊結構的高機率是澱粉樣蛋白形成的促成因素。然而,人們發現,自然界並不特別有利於 β 摺疊的形成,因為很少發現親水和疏水殘基序列模式的交替。
氨基酸序列的特徵會影響澱粉樣纖維結構和聚集速度。據稱,不同的突變,包括芳香族側鏈數量的變化、暴露表面積和偶極矩的變化,會改變許多多肽鏈的聚集速度。
未摺疊區域在促進部分摺疊蛋白質聚集中起著至關重要的作用。人們發現,一些易於彎曲或暴露於溶劑的區域喜歡聚集。其他未參與聚集的區域被發現沒有暴露,而是半埋藏,即使它們具有很高的聚集可能性,而結構中暴露且未參與聚集的區域則具有較低的聚集澱粉樣纖維的可能性。纖維傾向於透過未摺疊多肽片段的締合而聚集在一起,而不是透過對接結構元素。
總的來說,人們發現,未摺疊蛋白質的疏水性低於摺疊蛋白質,淨電荷高於摺疊蛋白質。傾向於不形成 β 摺疊結構蛋白質二級結構的殘基似乎會抑制澱粉樣蛋白聚集的發生。人們發現,蛋白質濃度、pH 值和離子強度與氨基酸序列有關,這會影響聚集速度。
環境影響
[edit | edit source]人們已經瞭解,蛋白質的一級結構(氨基酸序列)使蛋白質傾向於特定的三維結構,以及它如何從未摺疊形式摺疊到天然狀態。鹽濃度、溫度、主要溶劑的性質、大分子擁擠和伴侶蛋白的存在都會影響摺疊機制以及未摺疊蛋白質與天然狀態蛋白質的比例。最重要的是,這些環境因素會影響任何單個蛋白質達到正確最終結構的可能性。
放置在適當環境(特定溶劑、溶質濃度、pH 值、溫度等)中的孤立蛋白質傾向於“自摺疊”成正確的天然構象。改變任何這些環境特徵都會破壞結構和/或干擾摺疊機制。超出給定蛋白質“正常”範圍的 pH 值會使特定氨基酸電離或干擾原本會穩定結構的極性和偶極-偶極分子內力。過量的熱量(烹飪)蛋白質會破壞蛋白質二級結構所必需的氫鍵。
極端環境或化學變性劑的存在(如可以破壞二硫鍵的還原劑)會導致蛋白質變性並失去其二級和三級結構,形成“無規捲曲”。在某些條件下,完全變性的蛋白質可以恢復到其天然狀態。有意變性用於分析生物分子的各種方法中。
細胞內的複雜環境通常需要伴侶蛋白和其他生物分子,以便蛋白質正確形成天然狀態。
蛋白質是生命體必不可少的組成部分。人體發育需要與蛋白質的合成同步進行。然而,蛋白質包含著許多尚未解開的謎團,例如蛋白質摺疊。摺疊是蛋白質必要的活動,它們需要摺疊才能繼續發揮生物活性。摺疊也是每個蛋白質為了獲得穩定構象而經歷的過程。但有時這個過程會發生錯誤,科學家稱之為蛋白質錯誤摺疊。蛋白質錯誤摺疊會導致人類、動物和生物體出現多種疾病,例如阿爾茨海默病和瘋牛病。正因為如此,蛋白質摺疊和錯誤摺疊的研究變得至關重要。在揭示蛋白質、摺疊、錯誤摺疊及其影響的過程中,科學家們取得了許多成功,蛋白質的奧秘正逐漸被解開。科學家 W. A. (Bill) Thomasson 在文章《揭示蛋白質摺疊的奧秘》中記錄了許多關於蛋白質的重要內容;在這篇文章中,他闡述了阿爾茨海默病和瘋牛病,以及蛋白質錯誤摺疊的一些影響,並介紹了科學在這些領域取得的成果。Thomasson 博士首先從一般性的蛋白質摺疊和錯誤摺疊開始介紹。首先,蛋白質由氨基酸序列組成。科學家們已經發現了 20 種出現在蛋白質中的氨基酸。蛋白質結構以兩種基本形狀而聞名,即 α 螺旋和 β 摺疊。“大多數蛋白質可能在到達穩定構象的路上會經歷幾個中間狀態”(Campbell 和 Reece,79)。蛋白質需要摺疊才能繼續發揮活性。科學家們列出了三種蛋白質摺疊型別:蛋白質可以是摺疊的、部分摺疊的或錯誤摺疊的。在摺疊過程中,“被稱為伴侶蛋白的蛋白質與靶蛋白相關聯;然而,一旦摺疊完成(甚至之前),伴侶蛋白將離開其當前的蛋白質分子,繼續支援另一個蛋白質的摺疊”(Thomasson)。這篇文章的作者記錄了 Anfinsen 關於蛋白質錯誤摺疊的重要結論。在他看來,錯誤摺疊發生在摺疊過程中,當摺疊出錯時就會出現。蛋白質錯誤摺疊的研究集中在溫度敏感性突變上;科學家們觀察到噬菌體 P22 隨著溫度的變化而發生突變。他們得出結論,突變蛋白比正常蛋白穩定性差。這意味著,他們在噬菌體尾部突起的結論是,錯誤摺疊的蛋白質比正確摺疊的蛋白質穩定性差,而且它們很難達到正確摺疊的狀態。當蛋白質發生錯誤摺疊時,會導致許多疾病。聚集可能與錯誤摺疊的出現同時出現,並且它會出現在大腦中,導致阿爾茨海默病和瘋牛病,正如許多科學家認為的那樣。蛋白質錯誤摺疊對人類生活的一個影響就是阿爾茨海默病。這是一種老年病。根據科學家的研究,這種疾病發生在澱粉樣蛋白前體蛋白髮生錯誤摺疊時。這種蛋白質會被加工成可溶性肽 Aβ。科學家們還沒有完全瞭解這種疾病的起因。但導致錯誤摺疊的主要原因是血液中的載脂蛋白 E (apoE)。apoE 蛋白有三種形式,即 apoE2、apoE3 和 apoE4。每種形式的 apoE 對 Aβ 的影響尚未發現,但科學家認為 apoE 可以與 Aβ 結合。在錯誤摺疊過程中,β 澱粉樣蛋白形成,在阿爾茨海默病患者中形成“神經纖維纏結”。這種疾病只發生在老年人身上,因為在澱粉樣蛋白過程中,核的形成非常緩慢。這種蛋白質的突變不穩定,會導致疾病。關於 apoE 的研究仍然是一個謎,因為一些科學家表明,這種蛋白質的一種形式會導致疾病的發展,而另一種形式則會降低疾病的發展。最後,關於阿爾茨海默病的研究仍在繼續,目的是確認 apoE 蛋白對 Aβ 的影響,併成功找到治療這種疾病的方法。蛋白質錯誤摺疊的另一個影響是瘋牛病。這是一種非常危險的疾病,因為它可以從動物傳播到人類。這種疾病是由朊病毒的錯誤摺疊引起的。錯誤摺疊的過程是朊病毒的自我複製。朊病毒是含有 DNA 和 RNA 的蛋白質顆粒。突變出現在摺疊過程中,朊病毒自我複製並導致蛋白質錯誤摺疊。它們含有 DNA 和 RNA。這是蛋白質的一種特殊情況;它可以作為自己的伴侶蛋白。由於複製,朊病毒隨著正常蛋白質的增加而迅速繁殖。這種疾病表明,蛋白質摺疊可以在沒有遺傳學的情況下發生,例如在綿羊身上進行的實驗。Thomasson 博士在文章中繼續介紹了有關錯誤摺疊的更多資訊,以及科學家揭示其奧秘的方法。他提供了關於 p53 蛋白及其突變的資訊。它會導致癌症,也是一種蛋白質錯誤摺疊。Thomasson 博士想表達的是他關於藥物的想法,這種藥物可以使蛋白質錯誤摺疊變得更加穩定,並最大限度地減少蛋白質的錯誤摺疊。這個想法似乎很好,但它的結果就像蛋白質摺疊的奧秘一樣是一個謎。關於蛋白質摺疊的研究對我們的生活至關重要。錯誤摺疊是導致許多危險疾病的主要原因之一,但我們還沒有找到有效的治療方法。蛋白質摺疊的研究越來越成功,幫助人類能夠摧毀由錯誤摺疊引起的疾病。由蛋白質錯誤摺疊引起的疾病已經成為人類需要解決的問題之一。
分子伴侶主要以輔助蛋白質摺疊而聞名。伴侶蛋白不僅參與蛋白質生命週期的初始階段。分子伴侶參與生產、維持和回收蛋白質伴侶的結構和單元。伴侶蛋白存在於胞質溶膠中,但也存在於細胞隔室中,例如膜結合的線粒體和內質網。伴侶蛋白對蛋白質正確摺疊的作用或必要性各不相同。許多原核生物只有很少的伴侶蛋白,並且伴侶蛋白型別的冗餘度較低,而真核生物則有大量的伴侶蛋白家族,其中包含一些冗餘度。據推測,一些伴侶蛋白對於蛋白質的正確摺疊至關重要,例如原核生物的例子,它們具有較少的伴侶蛋白家族變異。其他伴侶蛋白髮揮的作用不那麼重要,例如在真核生物中,伴侶蛋白家族內部存在更多的變異,並且會產生效率或親和力的梯度。這種冗餘或低效伴侶蛋白的存在可能在一種狀態中存在,但伴侶蛋白的有效性也是其環境的函式。pH 值、空間、溫度、蛋白質聚集和其他外部因素可能會使原本無效的伴侶蛋白變成更重要的伴侶蛋白。這些環境因素表明了模擬細胞體內條件或天然狀態以瞭解需要使用伴侶蛋白的條件的重要性。這簡要概括了分析和比較伴侶蛋白體內與體外功能的困難。模擬體內或細胞內的環境不僅因為物理因素,如 pH 值或溫度,還因為伴侶蛋白開始使多肽構象化的時間。一些伴侶蛋白靠近核糖體,並立即附著到多肽上,以防止錯誤構象。其他伴侶蛋白允許多肽自己開始摺疊,並在稍後附著。因此,每個伴侶蛋白的作用變得與其靠近多肽的距離以及其輔助摺疊的時間和地點有關。最近的研究表明,核仁中的伴侶蛋白不僅催化蛋白質摺疊,而且還催化其他對維持健康細胞重要的功能。這些核仁伴侶蛋白被稱為核仁多功能蛋白 (NoMP)。例如,熱休克蛋白不僅幫助其他蛋白質摺疊,而且在壓力時刻發揮作用,以調節蛋白質穩態。此外,有證據表明,伴侶蛋白在網路中協同工作以監督某些功能,例如處理毒素、飢餓或感染。
核仁伴侶網路被劃分為不同的分支,每個分支都有其特定的功能。該網路是動態的,其網路組分的濃度或位置會隨著細胞生理和環境的變化而變化。熱休克蛋白 (HSP) 根據其分子量進行分類,是伴侶網路的組成部分。HSP 70s 和 90s 透過確保蛋白質正確摺疊並防止蛋白質毒性來維持蛋白質穩態,而蛋白質毒性是指由於蛋白質錯誤摺疊而導致的細胞功能損傷。HSP70s 幫助摺疊新合成的蛋白質,而 HSP90s 則在摺疊過程的後期發揮作用。核仁網路還包含參與核糖體生物發生或細胞中核糖體合成的伴侶。HSP70 和 DNAJ 家族中的蛋白質,它們幫助處理前 rRNA,經常在處理釀酒酵母 (一種酵母菌) 中的前 rRNA 的蛋白質複合物中被發現。其他 HSP 對於核糖體生物發生也很重要,包括 HSP90,它與 TAH1 和 PIH1 協同作用以建立小的核仁核糖核蛋白。核仁伴侶網路提供完成細胞存活所需的生物學任務所需的組織和幫助,如果它不能正常運作,可能會出現很多問題。例如,當癌細胞的 rRNA 合成水平升高時,核糖體生物發生就會增加。科學家正在研究化合物 CX-3543,該化合物可以阻止核仁與 rDNA 結合並阻止 RNA 合成,從而導致細胞死亡。有可能使用針對核仁伴侶網路中特定分支的藥物來治療功能失調的細胞。其他伴侶網路包括專門參與從頭蛋白質摺疊的網路,這意味著它們幫助摺疊新合成的蛋白質,以及對受損蛋白質的重新摺疊。在腫瘤細胞線粒體中存在的一個伴侶網路包含 HSP90 和 TRAP1,它們保護線粒體並防止細胞死亡,使癌細胞能夠繼續不受控制地擴散。[9]
示例:分子伴侶 (HSP 70)
[edit | edit source]HSP 70 是熱休克蛋白家族中的蛋白質,與 HSP 90 一起作用。它與 HSP 90 協同作用以支援蛋白質穩態。它在摺疊過程的早期與新合成的蛋白質結合。它具有三個主要結構域:N 末端 ATP 酶結構域、底物結合結構域和 C 末端結構域。N 末端 ATP 酶結合並水解 ATP,底物結合結構域對長達七個殘基的中性疏水性氨基酸殘基具有親和力,而 C 末端結構域充當底物結合結構域的蓋子。當 HSP 70 與 ATP 結合時,該蓋子處於開啟狀態,當 hsp 70 與 ADP 結合時,該蓋子處於關閉狀態。HSP70 或 DnaK 是細菌伴侶,可以透過夾緊肽段來幫助摺疊。[10]
-
 HSP 70
HSP 70

示例:GroEL 和 GroES
[edit | edit source]GroEL 和 GroES,或 60kDa 和 10kDa,都是細菌伴侶。GroEL 和 GroES 的結構都是堆疊的環,中間為空心。蛋白質適合在這個空心中心。然後,腔室內的構象變化可以改變蛋白質的形狀和摺疊。[10]
示例:分子伴侶 (HSP 90)
[edit | edit source]HSP 90 是熱休克蛋白家族中的蛋白質。然而,這種特殊的蛋白質與其他伴侶蛋白不同,因為 HSP90 在分子伴侶的摺疊方面受到限制。相反,Hsp 90 至關重要,需要對其進行研究和理解,因為許多癌細胞已經能夠接管並利用 Hsp 90 才能在許多毒性環境中生存。因此,如果有人對 Hsp90 抑制劑進行結構研究並以某種方式對其進行靶向治療,那麼就可能找到一種方法來阻止癌細胞的擴散。此外,已經進行了許多研究以測試 Hsp 90 伴侶迴圈是否是由 ATP 結合和水解驅動還是由其他因素驅動。但是,經過 Southworth 和 Agard 的大量研究,有足夠的證據表明 HSP90 蛋白可以在沒有核苷酸結合的情況下發生構象變化,而是平衡的穩定是將 Hsp90 改變為封閉狀態、緊湊狀態或開放狀態的因素。透過 X 射線晶體學以及單電子粒子顯微鏡,以及透過研究酵母 Hsp90、人 Hsp90 和細菌 Hsp 90 (HtpG) 的三態構象變化,發現 Hsp90 的三種構象,很明顯,特定物種存在不同的構象變化。總體而言,Hsp90 是一種伴侶蛋白,它更多地參與維持細胞內的穩態,而不是參與蛋白質摺疊。Hsp90 在未來藥物開發領域具有上升的潛力,因為它在幫助癌細胞存活方面發揮著至關重要的作用。
-
 HSP 90
HSP 90

示例:分子伴侶 (TF)
[edit | edit source]這是第一個與新鏈從核糖體通道出來時相互作用的伴侶蛋白。在沒有新鏈的情況下,TF 會迴圈開啟和關閉,但是一旦存在新鏈,它就會與新鏈結合,在新鏈周圍形成一個保護腔。為了發揮其功能,TF 會掃描新鏈的任何暴露的疏水性片段,並且它也可以重新與新鏈結合。在 TF 存在的情況下,摺疊效率更高,但是,這是以速度為代價的,它可以與新鏈保持 30 秒以上。當疏水性部分被掩蓋時,隨著摺疊向天然狀態進展,會觸發新鏈的釋放。
示例:分子伴侶 (YidC、Alb3、Oxa1)
[edit | edit source]YidC、Alb3 和 Oxa1 是促進蛋白質插入質膜的蛋白質。YidC 是一種僅包含兩個多肽鏈的蛋白質。其結構的形成得到了特定磷脂的支援。YidC 蛋白可以在革蘭氏陰性和革蘭氏陽性細菌中找到。Oxa1 可以線上粒體的內膜中找到。Alb3 位於葉綠體內部的類囊體膜中。實驗表明,YidC 蛋白積極地促進 Pf3 衣殼蛋白的插入。此外,YidC 還直接接觸 Pf3 衣殼蛋白的疏水性片段。雖然 Oxa1 只能在線粒體中找到,但它也可以促進核內膜蛋白的插入。YidC 和 Alb3 的作用似乎是可以互換的,因為 Alb3 可以替代 E. coli 中的 YidC。此外,YidC、Oxa1 和 Alb3 都支援 Sec 獨立蛋白質的插入。Oxa1 僅支援 Sec 獨立蛋白質的插入,因為酵母細胞中的線粒體沒有 Sec 蛋白。
NLR
[edit | edit source]富含亮氨酸的核苷酸結合域 (NLR) 提供了一種植物和動物中都存在的病原體感測機制。它們可以透過隱秘的機制直接或間接地由病原體分子衍生物觸發。研究表明,HSP90、SGT1 和 RAR1 等分子伴侶是 NLR 蛋白的主要穩定成分。HSP90 可以透過三種實用的方式監測其對應客戶(適用於 NLR 蛋白)的功能:促進功能閾值的穩態、啟用刺激依賴性活性以及提高進化能力。
植物包含許多 NLR 基因,這些基因被認為在 LRR 結構域中是多型性的,以便熟悉高度多樣化的病原體效應子。NLR 感測器的穩定性將是決定病原體識別的機制。HSP90 系統對植物有利,因為它會將亞穩態 NLR 蛋白耦合起來,並在訊號傳導能力強的條件下穩定它們。這將允許掩蓋可能造成損害的突變。
蛋白質摺疊中底物結合的分子伴侶機制
[edit | edit source]眾所周知,伴侶蛋白協同工作,幫助蛋白質摺疊,防止錯誤摺疊。然而,伴侶蛋白如何幫助蛋白質摺疊的機制尚未完全瞭解。最近關於Hsp40和Hsp70的研究為伴侶蛋白及其底物的機制提供了更多見解。Hsp40家族包含許多具有不同J結構域的Hsp40。當Hsp40與Hsp70結合時,不同的J結構域將執行不同的Hsp70 ATP酶活性。在蛋白質摺疊過程中,未摺疊的多肽與Hsp40伴侶蛋白結合。從那裡,Hsp40的J結構域與Hsp70的核苷酸結合域(NBD)結合。當HSP70 NBD上的ATP水解為ADP時,Hsp70底物結合域發生構象變化。這導致Hsp70對多肽底物的親和力更高,並從Hsp40上解離底物。當ADP被ATP交換時,多肽底物從Hsp40釋放。研究表明,核苷酸交換因子以降低Hsp70對ADP的親和力的方式改變了Hsp70 ATP酶域上的葉瓣。一旦多肽從Hsp70釋放,它就可以摺疊到其天然狀態,或者如果發生錯誤摺疊,它可以被伴侶蛋白重新摺疊。如果與Hsp70結合的多肽被E3泛素連線酶CHIP識別,它將被降解。[11]
小熱休克蛋白和α-晶狀體蛋白作為分子伴侶蛋白
[edit | edit source]眾所周知,小熱休克蛋白 (sHSPs) 和相關的 α-晶狀體蛋白 (αCs) 幾乎是無處不在的蛋白質,它們被各種壓力強烈誘導,但也構成性地在許多生物體的多種細胞型別中發揮作用。廣泛的研究表明,大多數 sHSPs 和 αCs 可以透過結合變性蛋白質和逆轉變性來充當 ATP 獨立的分子伴侶蛋白。這種方法從而保護細胞免受不可逆蛋白質聚集造成的損害。許多遺傳性疾病被發現是由 sHSP/αCs 中的缺陷引起的,這些蛋白質在神經退行性疾病和其他與蛋白質錯誤摺疊相關的疾病中積累。sHSP/αC 蛋白的大小範圍從 ~12 到 42 kDa,並且是位於 C 末端的約 90 個氨基酸的結構域,稱為 αC 結構域。sHSP-底物複合物可以透過尺寸排阻色譜觀察。它們很大且異質,並且它們的大小分佈取決於 sHSP/αC 與底物的比率以及底物聚集速率,底物聚集速率受濃度和溫度的影響。底物結合通常透過 sHSP/αC 上可用疏水錶面的增加來促進,這似乎在沒有明顯損失定義的 sHSP/αC 二級和三級結構的情況下發生。sHSP/αCs 上沒有單個特定的底物結合表面。相反,似乎許多位點有助於底物相互作用,並且結合可能因不同底物而異,具體取決於底物展開時暴露的表面的構象。然而,一些 sHSP/αCs 識別幾乎任何展開的蛋白質,這表明它們作用於任何不穩定或受損的細胞成分。
蛋白質摺疊的能量景觀
[edit | edit source]如果蛋白質隨機且不可預測地摺疊,則到達天然構象所需的時間將遠遠大於實際所需的時間。關於蛋白質如何自然有效地摺疊的當前理論涉及某種“漏斗”——這個想法是,存在著並非一步一步地達到正確的三維結構的方法,而是從上到下逐漸變窄的許多路徑。漏斗從頂部開始,從頂部能量不利摺疊向下進行,到底部能量有利於正確摺疊。
1961 年,克里斯蒂安·安芬森進行的實驗激發了蛋白質依靠能量學和熱力學來達到其天然摺疊的想法,當時他發現核糖核酸酶在被變性後可以自發地重新摺疊成其適當的結構,而無需其他分子的幫助。蛋白質摺疊不是隨機的這一理論的進一步理論證明見於萊文撒爾悖論,該悖論指出,一個長 100 個氨基酸的蛋白質大約需要 10^81 年才能達到正確的構象,而實際上,它只需要毫秒到一天的時間。

這些漏斗模型(如 Go 型模型)顯示出具有山丘和凸起的漏斗,代表蛋白質在能量漏斗中向下移動時走最小的阻力路徑。這些凸起被稱為“挫折點”。人們認為,具有最少挫折點或凸起的漏斗摺疊成其天然形式的速度更快,因為存在的能量邊界更少。雖然這些模型是簡化的嘗試,並沒有考慮到錯誤摺疊,但它們在許多蛋白質的情況下仍然證明是準確的。
另一個使用演算法和計算機的模型是經驗力場。該模型使用數十萬臺閒置執行的計算機以驚人的精度計算 50 個氨基酸以下的蛋白質摺疊場景。但是,這些計算機模型有時會高估不太可能的摺疊結構或產生很少或從未見過的摺疊模式。例如,一些模擬/演算法往往會卡在區域性最小值中,無法到達全域性最小值,即正確摺疊的蛋白質。Go 型模型等簡單模型不僅可以預測摺疊的蛋白質,還可以預測確定蛋白質摺疊速率的過渡狀態。
這些模型才剛剛開始展示蛋白質摺疊中間階段的動力學。因此,這是一個正在進一步研究的領域。對蛋白質摺疊動力學的理解還不太成熟,蛋白質在初始氨基酸鏈和最終產物之間的運動也是一個正在研究的領域。能量景觀模型也難以解釋擁擠和聚集等外部因素。一種這樣的外部相互作用的例子,稱為“多米諾骨牌交換”,涉及從一種蛋白質交換單體到另一種蛋白質,以啟用兩種蛋白質的正確摺疊。
最近的研究將人力和計算機能力結合起來,正確預測蛋白質構象。像 fold.it 這樣的網站,由華盛頓大學計算機科學系監督,將摺疊問題變成了影片遊戲,讓世界各地的人們像解謎遊戲一樣解決蛋白質摺疊問題。使用者被提供部分摺疊的蛋白質,通常是那些卡在區域性有利構象中,而這種構象對於計算機來說似乎是最優的,並被要求將蛋白質重新配置成看起來更穩定的形狀。利用計算機的計算能力和速度以及人類在空間中操縱物體的能力,在幫助更有效地解決蛋白質摺疊問題方面顯示出希望。
協同性和蛋白質摺疊速率
[edit | edit source]蛋白質摺疊中表達的協同性是蛋白質摺疊最引人注目的方面之一。與傳統的蛋白質摺疊涉及複雜和異質機制的觀點相反,許多蛋白質表現出的協同兩態摺疊動力學相對簡單。由於其簡單性,最近透過應用協同兩態摺疊動力學來理解決定協同性和蛋白質摺疊速率多樣性的努力。
蛋白質的協同性通常是指一種機制,在這種機制中,結構區域的存在使得在蛋白質摺疊中增加有序性更加有利。如前所述,小球狀蛋白質的協同兩態摺疊動力學相對簡單,成為許多科學家的研究興趣。對能夠對單個分子進行激發的敏感實驗,可以對過渡時間進行估計,揭示了兩種狀態的協同性。
由兩態摺疊蛋白質揭示的一般趨勢可以總結為以下兩點。首先,拓撲結構更復雜的蛋白質往往比拓撲結構更簡單、區域性的蛋白質摺疊得更慢;其次,更大的蛋白質往往比更小的蛋白質摺疊得更慢。這裡蛋白質的大小和大小是根據其鏈長定義的。
蛋白質摺疊動力學受過渡態能量增益和熵損失決定的自由能壘控制。在描述這種模式時,科學家引入了能量景觀理論的最小挫折原則。該理論指的是天然結構在蛋白質摺疊過程中比其他隨機構象具有更低的自由能的概念。因此,天然結構有利於蛋白質快速摺疊,並作為向天然狀態、蛋白質的功能形式或三級結構的驅動力。該原則可以用漏斗能量景觀來表達。
漏斗能量景觀
漏斗能量景觀將摺疊蛋白質的能量景觀描繪為一個粗糙的漏斗。粗糙來自蛋白質摺疊過程中的非天然接觸。該景觀本質上是多維的,因此漏斗是在二維圖上的投影。漏斗的深度表示構象狀態的能量;漏斗的寬度表示熵的度量。漏斗的瓶頸表示摺疊蛋白質的過渡態構象,而漏斗的底部表示蛋白質的天然狀態。當蛋白質走向其天然狀態時,它會經歷熵損失,並達到更低的能量狀態。漏斗能量景觀為科學家提供了方便的圖示,讓他們可以想象蛋白質摺疊過程的熱力學和動力學。
φ (phi) 值
另一個在蛋白質摺疊動力學研究中起作用的概念是φ (phi) 值。該值指的是過渡態構象中天然結構含量的近似測量。與 φ 值的比較是檢查研究蛋白質摺疊動力學的各種模型的方法之一。
一般觀察結果
第一個提到的趨勢可以從熵的角度很容易理解。拓撲結構更復雜的蛋白質,或具有長程相互作用的蛋白質,與具有短程相互作用的蛋白質相比,在摺疊方面的熵成本預計會更高。第二種趨勢最近透過專注於蛋白質大小對摺疊速率影響的實驗得到了證實。研究發現,僅基於鏈長的簡單模型可以粗略地預測蛋白質的摺疊速率和穩定性。
Go¯模型
粗粒度拓撲模型(Go¯模型)被廣泛用於研究蛋白質摺疊的協同性和動力學,因為人們注意到天然蛋白質的拓撲結構決定了摺疊機制。典型的 Go¯模型簡化了蛋白質,其中只有一種相互作用穩定摺疊的蛋白質。早期的模型經常檢查蛋白質摺疊中起作用的非加性力,例如側鏈排序和疏水效應。近年來,更多種類的 Go¯模型被用於研究蛋白質摺疊動力學。
- 專案符號列表項
- 具有蛋白質天然接觸之間非成對加性相互作用的 Go¯模型(這裡指的是 Eastwood 和 Wolynes 的模型)表明,短程多體相互作用可以增加自由能壘,並使過渡態構型更加區域性化。
- 另一方面,晶格 Go¯模型表明,區域性和核心埋藏相互作用的耦合促進了協同性,並增加了與接觸順序的相關性。
- 具有成對加性相互作用的 Go¯模型,特別是那些專注於三體相互作用強度變化的影響和 φ 值的模型,表明三體相互作用增加了能壘,並增加了與測量的 φ 值的一致性。
- 此外,溶劑介導的相互作用也被引入 Go¯模型。當接觸之間的相互作用被溶劑分離的最小值和脫溶能壘取代時,觀察到蛋白質功能的動力學和協同性隨著脫溶能壘高度的增加而增加。溶劑介導的 Go¯模型的優點是它有助於區分短程接觸和長程接觸,從而區分具有簡單拓撲結構的蛋白質和具有更復雜拓撲結構的蛋白質。在溶劑介導的 Go¯模型的研究中,經常使用“化學計量圖”。化學計量圖是一種用不同濃度的變性劑來表示蛋白質摺疊動力學資料的圖形,這種變性劑會破壞蛋白質的天然結構。
- 變分 Go¯模型透過排除處於天然狀態下緊密接觸的殘基之間的體積力來提高協同性。在這個模型中,實現以下目標:a)長程接觸的協同性更強;b)計算速率的範圍更廣;c)計算的 φ 值得到改善。還有一些 Go¯模型完全專注於蛋白質摺疊能量景觀的漏斗方面,而忽略了非天然接觸的影響。
其他模型,例如毛細管模型,假設摺疊核的體積與單體的數量成比例。在這種模型中,表明協同性增加往往會減慢動力學並使能量景觀平滑。
結論。
近年來,具有非加性力的拓撲模型的發展正在成為理解蛋白質摺疊速率協同性的更流行和可靠的方法。該模型的改進顯示出其在更明確和深入地理解決定蛋白質摺疊速率和機制方面的光明前景。能夠實現長程接觸的 Go¯模型變得更加協同,φ 值更加準確,需要在蛋白質摺疊動力學和摺疊機制的研究中得到進一步改進和更多關注。
蛋白質序列、結構和功能之間的關係
[edit | edit source]在過去的 20 年中,人們開發了幾種蛋白質預測方法。還沒有開發出適用於所有蛋白質的通用方法,因為每種方法都有其優點和缺點。開發這種方法的難度在於我們對蛋白質序列、結構和功能之間高度錯綜複雜的關係的理解還不夠完善。
Anfinsen 提出了將氨基酸序列與其結構相關聯的理論。他證明了一種變性(未摺疊)蛋白質可以自發地恢復其天然三級結構。這種方法也是將功能分配給蛋白質結構的有用貢獻者。蛋白質研究人員可以預測疏水底物可能與蛋白質的疏水區域結合,反之亦然,對於帶電區域也是如此。這種方法的問題是,它沒有考慮某些因素,例如非典型環境條件。
人們認為,相似的排序意味著相關的結構。這種理論只對少數蛋白質適用。研究人員發現,蛋白質摺疊的相似性並不總是與其蛋白質序列相關。由於這些發現,1995 年提出了“帕拉塞爾蘇斯挑戰”。“帕拉塞爾蘇斯挑戰”背後的理論是開發兩種蛋白質,它們的序列相似性超過 50%,但它們的摺疊完全不同。該挑戰在 1997 年得到了滿足,當時出現了兩個共享 88% 序列同一性(GA88 和 GB88)的蛋白質序列。最近的研究表明,只要 3 個突變就足以誘導不同的摺疊模式。儘管“帕拉塞爾蘇斯挑戰”的結果非常有趣,但它們很少在自然界中發生。
功能收斂導致將特定功能分配給結構的問題。各種結構可以採用相似的功能,但有些結構也可以採用非常不同的功能。但是,某些摺疊與特定功能之間存在顯著相關性。功能預測中有兩個主要變數:(1)結合位點的位置,以及(2)位點上的功能範圍。對功能有貢獻的金屬、離子、輔助因子和其他蛋白質也必須考慮在內。在透過晶體學確定結構時,會遇到一個問題。PROCOGNATE 資源和 PIDA 資料庫為這個問題提供了一個解決方案。
定義蛋白質功能的一種廣泛使用的方法源自基因本體,它包含三個圖結構,其中定義了功能術語和它們之間的關係。基因本體的侷限性出現在非位置蛋白質,以及當蛋白質在晶體結構中沒有與配體之間定義的關係時。試圖彌合這一差距的其他發展包括蛋白質特徵本體 (PFO [29]) 和分散式註釋系統 (DAS[30])。
兩種方法用於確定功能位點:(1)在不知道位點在哪裡或它結合什麼的情況下,或者(2)在事先知道相互作用夥伴的情況下。最常用的方法涉及生物資訊學,例如 SOIPPA 方法。序列保守性是將功能分配給蛋白質的一個非常重要的貢獻者,但很難確定殘基是由於結構原因還是功能原因而保守的。另一種方法涉及基於能量的方法。最近開發的 ProFunc 伺服器結合了 InterProScan 和 BLAST 搜尋等方法。
預測結合位點(本身極其複雜)只是拼圖的第一步。下一步是根據生化功能確定整體功能,更具挑戰性的是確定其生物學作用。當研究人員發現蛋白質功能不僅取決於其最終摺疊產物時,分析蛋白質功能的難度增加了另一層複雜性。蛋白質在其部分變性和完全變性狀態下可能具有功能。綜上所述,可以肯定地說,我們還有很多關於蛋白質序列、結構和功能之間關係的知識需要學習。
結構域交換、摺疊和錯誤摺疊
[edit | edit source]蛋白質中發生的結構域交換可能在蛋白質的摺疊或錯誤摺疊過程中很重要。結構域交換髮生在兩個或多個相同的蛋白質鏈相互交換時。結構域交換可以被認為是單體和寡聚體交換的一種機制。寡聚體交換中發生的是,來自一種蛋白質的一個單體將與來自另一種蛋白質的另一個相同的單體交換。這種結構域交換機制已在各種蛋白質中觀察到,超過 40 種不同的蛋白質。交換機制對某些蛋白質的功能很重要。例如,對於特定蛋白質 p13suc1,人們已經觀察到交換和聚集是相關的,這意味著它們具有共同的機制。P13suc1 在細胞週期程序中是細胞週期蛋白依賴性激酶 (Cdk) 所必需的。P13suc1 具有兩種不同的狀態,一種是單體,另一種是交換的二聚體。結構域交換部分是一個 β 鏈,它不是一個獨立摺疊的結構域。在研究過程中,人們發現 β4 在與 β2 接觸時起著至關重要的作用,因為它們在摺疊過程的早期階段相互配對。因此,對於 p13suc1,已經證明,已交換的區域負責蛋白質的摺疊和錯誤摺疊。蛋白質中似乎存在摺疊和錯誤摺疊之間的競爭,因為多肽鏈可以摺疊成結構或錯誤摺疊成澱粉樣纖維。在蛋白質摺疊中,更重要的是摺疊核的存在,摺疊核在過渡態中形成蛋白質鏈的一部分。人們還發現了參與蛋白質摺疊核位置的殘基與澱粉樣蛋白區域之間的相關性,以及纖維形成和蛋白質摺疊可能包含關鍵殘基的重要資訊。透過對蛋白質摺疊進行建模並觀察寡聚形式中的可交換區域,可以看到這種關係是摺疊和錯誤摺疊的原因。這可能使研究人員更接近解決蛋白質求解問題並瞭解蛋白質是如何獲得其摺疊指令的。參考資料:http://www.benthamscience.com/open/tobiocj/articles/V005/27TOBIOCJ.pdf
-
 結構域交換
結構域交換

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
死亡摺疊超家族包含 4 種亞家族結構。它們分別是死亡結構域 (DD)、死亡效應結構域 (DED)、半胱天冬酶募集結構域 (CARD) 和 PYrin 結構域 (PYD)。這些亞家族結構參與多聚體複合物的組裝,這些複合物可能與細胞炎症和死亡有關。
死亡摺疊結構域的結構和功能
目前已知有 102 種蛋白質具有死亡摺疊超家族結構域。這些結構域包含同型相互作用。這些蛋白質包括 39 個 DD、8 個 DED、33 個 CARD 和 22 個 PYD。儘管這些結構域在序列上最多有 90% 的差異,但它們都具有典型的死亡摺疊。這種摺疊“包含一個球狀結構,其中 6 個兩親α螺旋以反平行α螺旋束的形式排列,具有希臘鍵拓撲結構”(Peter Vandenabeele 等人,2012 年)。構成這些亞家族之一的死亡結構域之間的差異在於 α 螺旋的長度和方向以及疏水殘基和帶電殘基在複合物表面上的分佈。
死亡摺疊結構域的推測功能是介導大型寡聚訊號複合物的組裝。在這些複合物中,半胱天冬酶和激酶的活性增加。在此之前,人們對具有死亡摺疊結構域的蛋白質組裝體的結構構象知之甚少。
三種不同的相互作用型別
I 型相互作用:來自一個死亡摺疊結構域的螺旋 1 和 4(Patch Ia)的殘基與來自另一個死亡摺疊結構域的螺旋 2 和 3(Patch Ib)的殘基相互作用。II 型相互作用:來自一個死亡摺疊結構域的螺旋 4 和螺旋 4 和 5 之間的環(Patch IIa)的殘基與來自另一個死亡摺疊結構域的螺旋 5 和 6 之間的環(Patch IIb)的殘基相互作用。III 型相互作用:來自一個死亡摺疊結構域的螺旋 3(Patch IIIa)的殘基與位於另一個死亡摺疊結構域的螺旋 1 和 2 之間的環以及螺旋 3 和 4 之間的環(Patch IIIb)的殘基相互作用。
先前的理論認為,這三種相互作用型別在整個死亡摺疊超家族中是保守的,但現在看來,相同型別死亡摺疊結構域之間的相互作用存在差異。
死亡摺疊結構域的晶體分析
只有三種 DD 複合物的晶體結構被分析過。它們是 PIDDosome、MyDDosome 和 Fas/FADD-DISC。對這些結構的分析表明,DD 可以參與多達六種相互作用。
死亡結構域和藥物
死亡結構域已被證明可以促進導致炎症和細胞死亡的多聚體複合物的組裝。瞭解這些結構可以透過阻止或觸發這些寡聚體複合物的形成,從而產生治療益處。可能受這些相互作用影響的疾病包括神經退行性疾病和炎症性疾病,以及許多其他具有炎症或過度細胞死亡特徵的疾病。
雖然摺疊通常是蛋白質功能的主要貢獻者,但有些蛋白質並不摺疊成特定的結構,但仍然具有功能。這些蛋白質通常在不同的形式之間轉換,或者具有不保持特定形狀的無序區域,而不是特定的結構。
正如蛋白質的摺疊由其氨基酸序列決定一樣,非摺疊蛋白質之所以是非摺疊的,也是因為它們的序列。這些蛋白質的某些氨基酸含量往往比摺疊蛋白質少得多,而另一些氨基酸則多得多。具體來說,非摺疊蛋白的形成疏水核心的氨基酸含量較少,而表面氨基酸含量較多。疏水核心的形成是大多數蛋白質摺疊的第一步,一旦形成,核心往往會提供穩定最終結構的驅動力。如果沒有形成核心的氨基酸,蛋白質就不會被驅動到特定的結構。
一些在非壓力條件下完全摺疊且不活躍的分子伴侶被稱為條件性無序蛋白。當這些伴侶暴露於不同的壓力條件下時,它們會呈現部分無序的構象。這種無序非常重要,因為它們能夠保護細胞免受壓力源的影響。對這些無序伴侶的研究使人們對蛋白質無序在分子識別中的功能作用有了更多瞭解。X 射線晶體學是一種有用的技術,有助於視覺化蛋白質的結構。根據這種技術,超過 95% 的整個分子由 25% 的晶體結構表示,而所有其他分子由於這些區域上的多種構象,其序列中超過 5% 的電子密度缺失。蛋白質實際上具有一些無序構象,這些無序蛋白在連續光譜上從非常靈活的結構狀態到靜態結構狀態的一端。這種無序中可能只有一部分蛋白質或整個完整的多肽鏈。因此,僅研究蛋白質的某些部分並不能幫助總結蛋白質的靈活性。“條件性無序”一詞表示蛋白質的無序可能在某些特定條件下發生,而在其他條件下可能不會發生。在蛋白質中看到內在無序的情況非常普遍。例如,估計真核蛋白質的 30% 到 50% 具有超過 30 個氨基酸,這些氨基酸在體外違反了已定義的二級結構,並且已經預測存在許多完整的無結構蛋白質。儘管使用了許多計算方法,但仍然很難驗證細胞內蛋白質摺疊狀態。許多蛋白質在細胞內部分或完全摺疊,而實際上在細胞內是無結構的。這些機會的數量仍然不確定。然而,人們認為,適當的結合對的存在將使無序蛋白進入其摺疊狀態,這意味著體外固有無序蛋白的百分比在細胞內可能更低。細胞內的穩定相互作用可能會降低無序的程度。透過化學位移、殘餘偶極偶合和順磁共振增強測量,NMR 成為提供有關蛋白質無序程度的詳細資訊的一種好方法。
無序蛋白有兩種狀態。一種表現出高度的靈活性,而另一種狀態是蛋白質表現出更多有序的狀態。因此,為了瞭解無序和功能之間的因果關係,研究這兩種狀態至關重要。許多無序蛋白,如 DNA、蛋白質和膜,一旦找到結合夥伴就會重新摺疊。此外,還可以發生有序到無序到有序的轉變。參與多種結合的蛋白質是條件性無序的很好的例子。與已經井然有序的結合表面相比,在結合之前無序的結合表面能夠更好地摺疊成與其他夥伴不同的構象。“構象選擇假說”表明,構象集合的不同成員可以透過不同夥伴的結合而穩定。另一方面,“結合摺疊”模型提出,蛋白質在與不同夥伴結合時可能能夠摺疊成不同的構象。
對整個蛋白質組的預測表明,真核生物中無序蛋白的頻率遠大於原核生物,而兩組原核生物,古細菌和細菌,的頻率相似。在哺乳動物中,大約一半的蛋白質被預測具有大型無序區域,其中大約四分之一是完全無序的。
無序蛋白在訊號傳導和調控中普遍存在,特別是在與生物分子(如核酸和其他蛋白質)的相互作用中。分子識別、蛋白質組裝和修飾通常涉及具有無序區域的蛋白質。這些蛋白質能夠與多個分子伴侶相互作用,這意味著它們在蛋白質-蛋白質網路中也很常見,要麼作為樞紐蛋白,要麼作為與樞紐蛋白相互作用的蛋白質。
無序蛋白與多種人類疾病有關。特別是澱粉樣蛋白疾病,涉及錯誤摺疊蛋白質的積累,似乎與無序蛋白有關,這可能是因為它們的變異區域使其更有可能具有有利於其積累的結構。這一類別包括許多神經退行性疾病,如阿爾茨海默病和帕金森病。
可以使用計算機演算法透過氨基酸的一級結構或氨基酸序列來分析和比較氨基酸的結構或摺疊,以及由此延伸的功能。使用計算機可以增強對未知摺疊模式的氨基酸序列與具有相似氨基酸序列的已知摺疊的比較。一種名為蛋白質基本區域性比對搜尋工具,或 蛋白質 BLAST 的計算機自動化工具,是一個向公眾開放的免費搜尋工具,允許線上資料庫中快速比較氨基酸序列。此工具的輸出是氨基酸的匹配百分比和序列匹配的已知特性。此外,由於氨基酸序列基於 DNA 序列,三個鹼基編碼一個氨基酸,因此可以使用 DNA BLAST 在 DNA 水平上分析所研究的蛋白質。公共氨基酸和 DNA 序列資料庫的整合以及計算機演算法的應用,透過允許世界各地的科學家共享和分析序列,加速了基因組和蛋白質組領域的發展。
附錄
計算機的作用
建立 BLAST 程式的科學家是來自 NIH 的 Webb Miller、David J. Lipman、Warren Gish、Eugene Myers 和 Stephen Altschul
分子伴侶
Pain, Roger H. 蛋白質摺疊機制。第 2 版。364-85
蛋白質摺疊的能量景觀
Cho, Samuel S. “蛋白質摺疊、結合和聚集的能量景觀:簡單漏斗及超越”。加州大學聖地亞哥分校博士論文(2007)。
Cheung, Margaret S. “與分子功能相關的蛋白質摺疊動力學的能量景觀方面”。加州大學聖地亞哥分校博士論文(2003)。
Yang, Sichun. “擴充套件蛋白質摺疊動力學理論框架”。加州大學聖地亞哥分校博士論文(2006)。
分子內相互作用
Pain, Roger H. “蛋白質摺疊機制” 第 2 版。
http://www.nature.com/horizon/proteinfolding/background/importance.html
Berg “生物化學” 第 6 版
計算機模擬研究幫助確定了共翻譯摺疊途徑的幾個特徵。首先,確定了體內蛋白質摺疊是一個向量過程,這是一個分散變化。其次,從 N 端到 C 端發育中的多肽的共翻譯向量摺疊導致從核糖體通道中出現的多肽不同區域的順序結構。第三,在蛋白質合成過程中,發育中的多肽鏈與核糖體的連線減少了生長鏈的構象空間和自由度。這限制了可能的中間體的數量,並減少了可能的摺疊途徑的數量。第四,共翻譯蛋白質摺疊在核糖體上多肽鏈合成過程的早期開始,一些元素在核糖體通道內形成。第五,摺疊催化和分子伴侶在生長鏈從通道中出現後立即與其相互作用。這加速了蛋白質摺疊中的緩慢步驟,並防止蛋白質錯誤摺疊。
- ↑ “阿爾茨海默病”. 俄亥俄州立大學醫療中心. 2009. 檢索於 2009-10-09.
{{cite web}}: 外部連結位於|publisher= - ↑ Lindquist, Susan (1999). “什麼是朊病毒?”. 檢索於 2009-10-09.
- ↑ “瘋牛病和變異型克雅氏病”. eMedicine 健康. 檢索於 2009-10-09.
{{cite web}}: 外部連結位於|publisher= - ↑ “揭開蛋白質摺疊之謎” (PDF). [Thomasson, W.A. “揭開蛋白質摺疊之謎”]. 檢索於 2009-10-18.
- ↑ “摺疊解決囊性纖維化”. [1]. 檢索於 2009-10-18.
{{cite web}}: 外部連結位於|publisher= - ↑ “遺傳疾病概況:鐮狀細胞貧血”. [2]. 檢索於 2009-10-18.
{{cite web}}: 外部連結位於|publisher= - ↑ “亨廷頓舞蹈症基礎”. [3]. 檢索於 2009-10-18.
{{cite web}}: 外部連結位於|publisher= - ↑ a b c 無效的
<ref>標籤;未為名為annu的引用提供文字 - ↑ Piotr Banski, Mohamed Kodiha and Ursula Stochaj (2010). “核仁中的伴侶蛋白和多工蛋白:為了生存而相互連線?”. 檢索於 2010-10-16.
- ↑ a b Joan L. Slonczewski, John W. Foster. "Microbiology: An Evolving Science."
- ↑ Summers, Daniel W., and Peter M. Dougla (2009). "Polypeptide Transfer from Hsp40 to Hsp70 Molecular Chaperones". Retrieved 2010-10-24.
{{cite web}}: CS1 maint: multiple names: authors list (link) - ↑ 11. Kersse K, Verspurten J, Vanden Berghe T, Vandenabeele P. The death-fold superfamily of homotypic interaction motifs. Trends in biochemical sciences. 2011;36(10):541–52. Available at: http://www.ncbi.nlm.nih.gov/pubmed/21798745. Accessed October 29, 2012.
12. Small heat shock proteins and α-crystallins: dynamic proteins with flexible functions. Basha E, O'Neill H, Vierling E. Trends Biochem Sci. 2012 Mar;37(3):106-17. Epub 2011 Dec 14
Conditional disorder in chaperone action. Bardwell JC, Jakob U. Trends Biochem Sci. 2012 Sep 24. pii: S0968-0004(12)00127-2. doi: 10.1016/j.tibs.2012.08.006. [Epub PMID 23018052 [PubMed - as supplied by publisher] "Molecular Biology of the cell." Fifth Ed-Alberts, Johnson, Lewis, Raff, Roberts, Walter. pg. 716-717
Braakman, Ineke, and Neil J. Bulleid. "Protein Folding and Modification in the Mammalian Endoplasmic Reticulum." Annual Review of Biochemistry. 80. (2011): 71-99. Web. 29 Oct. 2011. <http://www.annualreviews.org/doi/pdf/10.1146/annurev-biochem-062209-093836>.
Cabrita LD, Dobson CM, Christodoulou J. Protein folding on the ribosome. Current Opinion in Structural Biology 2010, doi:10.1016/j.sbi.2010.01.005
A Keith Dunker, Israel Silman, Vladimir N Uversky, Joel L Sussman. "Function and structure of inherently disordered protein." Curr Opin Struct Biol. 2008 Dec;18(6):756-64
Booth Paula J, Curnow Paul. Folding Scene Investigation: Membrane Proteins. Current Opinion in Structural Biology 2009, doi:10.1016/j.sbi.2008.12.005
Heijne, Gunnar Von. "Membrane Protein Folding and Insertion." Annual Review of Biochemistry 80 (2011): 157-160. 26 Oct. 2011 <http://www.annualreviews.org/doi/full/10.1146/annurev-biochem-111910-091345?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%3dpubmed>
Kuhn, Andreas, Rosemary Stuart, Ralph Henry, and Ross E. Dalbey. "The Alb3/Oxa1/YidC protein family: membrane-localized chaperones facilitating membrane protein insertion?" TRENDS in Cell Biology 13 (2003): 510-516. 26 Oct. 2011 <http://www.cell.com/trends/cell-biology/abstract/S0962-8924(03)00196-X>
表 1:Berg,Jeremy。二級結構中氨基酸殘基的相對頻率。2012。生物化學,紐約。印刷。
Voet,Donald,Judith G. Voet。生物化學第三版。新澤西州:John Wiley & Sons,Inc,2004。印刷。